Health Disparities in Pathology Education: An Examination of the 10th Edition of Robbins Basic Pathology
Last Updated: February 21, 2025
Andrea T. Deyrup, M.D., Ph.D.
This document came to life as an attempt to provide context for the medical students I was teaching using the 10th edition of Robbins Basic Pathology, before the 11th edition was published. I used this text for the 6 years since it was published and was troubled by the associations made between race and disease incidence and prognosis. As a co-editor of the 11th edition of the textbook, I worked with my colleague, Dr. Joseph Graves, Jr., and the other editors to remove race-based medicine from the text. Although the new edition is much improved, it is worth sharing this information since generations of clinicians (including the attendings and residents you’ll be working with!) have been taught these associations.
I began by searching through the electronic version of the 10th edition to identify race-based context. This search yielded more than 35 diseases for which health disparities were associated with race, ethnicity or ancestry. I then did a deep dive into the literature to determine the validity and accuracy of these statements.
I would like to acknowledge Dr. Joseph Graves, Jr., an evolutionary biologist at North Carolina Agriculture and Technical State University, the largest historically black university in the country. Dr. Graves has read over this document and provided relevant suggestions and recommendations. He is a valued colleague and comrade in arms.
A few thoughts about “race” in pathology
Race is a social/political construct. It is more meaningful, in the context of pathophysiology, to focus on geographic ancestry. For instance, populations that evolved under pressure from Plasmodium falciparum malaria (e.g., sub-Saharan Africa, parts of Asia, the Middle East and Mediterranean Europe) have an increased incidence of sickle cell disease since sickling promotes clearance of infected red blood cells.
A second influence on health disparities relates to the “founder effect” either due to a population bottleneck (e.g., Finland, which is known for “Finnish heritage diseases”) or social norms (e.g., endogamy, marriage within one’s community). Founder effects are thought to be responsible for a wide range of recessive diseases and conditions including Tay-Sachs disease in Ashkenazi Jews, intolerance of certain anesthetics among people with Vysya ancestry in India, and an increased incidence of ankylosing spondylitis in people with the surname Reddy (Nakatsuka N et al, 2017).
Letting go of the idea of race as a meaningful diagnostic parameter has been challenging for many physicians. The scientific literature is replete with papers that correlate this or that disease/condition/prognosis with what we now know to be an inaccurate and often meaningless racial identity. However, many physicians fear that by discarding racial “data”, we are letting political correctness impede clinical care.
We must remember that “racial profiling” in medicine harms everyone. A blonde, blue-eyed man may suffer for years only to have a genome test reveal he has familial Mediterranean fever. An overweight indigenous woman presenting with right upper quadrant pain may be labeled as “gallstones” and an ectopic pregnancy overlooked. A girl of African descent may not be diagnosed with cystic fibrosis until she is 8 years old. We must always question all of the data. All of the time.
Nakatsuka N, Moorjani P, Rai N, Sarkar B, Tandon A, Patterson N, Bhavani GS, Girisha KM, Mustak MS, Srinivasan S, Kaushik A, Vahab SA, Jagadeesh SM, Satyamoorthy K, Singh L, Reich D, Thangaraj K. The promise of discovering population-specific disease-associated genes in South Asia. Nat Genet. 2017 Sep;49(9):1403-1407. doi: 10.1038/ng.3917. Epub 2017 Jul 17. PMID: 28714977; PMCID: PMC5675555.
A few notes about “race” in scientific studies
Humans do not have biological races; therefore, much of the literature on racial differences must be viewed with a skeptical eye. For one thing, it is important to determine how the study determined race: was it self-reported by the patient or was it an administrator/researcher making his/her own best guess based on appearances or last name? Neither of these methods is particularly accurate.
In any case, what these methods are determining is a socially defined category, not a natural biologically determined demarcation in the human species. Thus, what most people think of as race is deeply flawed. In modern biology, races are defined by two criteria: the amount of genetic variation within and between groups, or whether a group can be determined as a unique evolutionary lineage by phylogenetic methods. It has long been known that humans show more variability within so called “races” than there is between them. Neither is there any evidence that any group of humans can be viewed as representing a unique evolutionary lineage (Rosenberg et al. 2005; Lawon-Handley et al. 2007).
In working through the scientific literature, I am using the terminology that was employed in the original studies. You will see terms that, in many respects, are essentially meaningless such as “Asian” (Asia refers to a land mass that extends from Turkey to Southern Russia to Japan to Indonesia and includes China, India and the countries of the Middle East). How can one possibly say a disease is more common in Asians?!?
I will replace “Caucasian,” which is an 18th century and inaccurate term that is related to scientific racism. Originally this term included Northern Indians but now its closest approximation has come to mean individuals of Northern European descent/European descent, Whites, and White Americans.
Chapter 2: Inflammation
Keloids
“Certain individuals seem to be predisposed to keloid formation, particularly those of African descent.” (BP10, p 93)
Keloids are benign fibroblastic proliferations that arise in the setting of skin injury in genetically susceptible individuals. Unlike hypertrophic scars, they can extend past the boundaries of the original wound and do not improve with time. Histologically, there are dense collagen bundles in the dermis, roughly parallel to the skin surface. The mechanism of keloid formation is unknown; however, it is likely that there are imbalances/alterations in growth factors (especially transforming growth factor b) and other factors involved in wound repair (e.g., matrix metalloproteinases and other extracellular matrix-degrading enzymes).
Although it is commonly stated in the literature that keloids are more common in people of African, Asian and, to a lesser degree, Hispanic and Mediterranean descent, the figures in the literature are filled with misinformation and misquotations. For example, the “fact” that keloids are 15 times more likely in dark skinned individuals (Chike-Obi CJ, 2009; Robles DT and Berg D, 2007; Brissett AE and Sherris DA, 2001; Gao F-L et al, 2005; Huang C et al, 2013) originates from a 1969 paper that actually says that “the relatively fair-skinned Chinese appear to be slightly more prone to keloids than the dark-skinned Indians and Malays.” (Alhady SM and Sivanantharajah K, 1969). Looking at the actual data, it appears that the lighter skinned patients were 2.4 to 3.3 times more likely than the darker skinned patients to develop keloids.
In UpToDate (accessed 11/21/21), the following association was made: “Keloids have been reported in 5 to 16 percent of individuals of Hispanic and African ancestry”. I only found the origin of this “16%” following a VERY deep dive into the literature, reviewing more than 30 articles back to 1931. I recorded a video which I then sent to the authors/editors of this section of UpToDate. As of 12/23/21, UpToDate has now removed the association of race/ethnicity and keloids in the epidemiology section! They have also removed the race-based material from the Genetics session following an article by Dr. Graves and myself in the New England Journal of Medicine.
My video was also used to change recommendations for intradermal vs subcutaneous Mpox vaccinations by the California Department of Public Health.
Another “fact” in the literature is that: “patients who suffer from albinism rarely develop keloids” (Gao F-L et al, 2005). This datum is often cited to support a higher incidence of keloids in darker skinned individuals; however, this information, too, is not supported by rigorous science: a population-based study in Africa by Kiprono et al (2015) found an equal incidence of keloids in patients with and without albinism.
The tendency towards keloid formation is believed to have a genetic component based on correlation with family history, twin concordance and cases of familial keloids (Kiprono SK et al, 2015; Ramakrishnan KM et al, 1974; Marneros AG 2001). A role for melanocytes has been implicated (Gao F-L et al, 2005); however, this study is also one that 1) denied keloids in patients with albinism and 2) perpetuated the statistic of a 15-fold increase in keloids in darker skinned patients. Gene-based association studies are currently being performed in order to determine more clearly the pathogenesis since this has implications for treatment (Hellwege, J.N. et al, 2018; Glass, R.A., 2018).
What is the clinical significance of all this? Keloid formation is of clinical concern since patients at risk for keloid formation may delay or avoid necessary surgery, particularly of the head and neck, for cosmetic reasons. A plastic surgeon of African descent, Ivens LeFlore (1980), has a very matter of fact approach: Rather than look at the color of the patient’s skin and make an assessment of the patient’s risk of developing keloids… look at the patient’s skin! If an individual has a tendency to keloid formation, by the time s/he reaches adulthood, there should be evidence of this propensity. Treatment decisions can be based on THAT patient’s risk, not on misconceptions related to incomplete research, inaccurate citations and a false concept of biological race.
Alhady SM, Sivanantharajah K. Keloids in various races. A review of 175 cases. Plast Reconstr Surg. 1969 Dec;44(6):564-6. doi: 10.1097/00006534-196912000-00006. PMID: 5352921.
Brissett AE, Sherris DA. Scar contractures, hypertrophic scars, and keloids. Facial Plast Surg. 2001 Nov;17(4):263-72. doi: 10.1055/s-2001-18827. PMID: 11735059.
Chike-Obi CJ, Cole PD, Brissett AE. Keloids: pathogenesis, clinical features, and management. Semin Plast Surg. 2009 Aug;23(3):178-84. doi: 10.1055/s-0029-1224797. PMID: 20676312; PMCID: PMC2884925.
Deyrup A, Graves JL Jr. Racial Biology and Medical Misconceptions. N Engl J Med. 2022 Feb 10;386(6):501-503. doi: 10.1056/NEJMp2116224. Epub 2022 Feb 5. PMID: 35119803.
Gao FL, Jin R, Zhang L, Zhang YG. The contribution of melanocytes to pathological scar formation during wound healing. Int J Clin Exp Med. 2013 Aug 1;6(7):609-13. PMID: 23936604; PMCID: PMC3731197.
Glass DA 2nd. Current Understanding of the Genetic Causes of Keloid Formation. J Investig Dermatol Symp Proc. 2017 Oct;18(2):S50-S53. doi: 10.1016/j.jisp.2016.10.024. PMID: 28941494.
Hellwege JN, Russell SB, Williams SM, Edwards TL, Velez Edwards DR. Gene-based evaluation of low-frequency variation and genetically-predicted gene expression impacting risk of keloid formation. Ann Hum Genet. 2018 Jul;82(4):206-215. doi: 10.1111/ahg.12245. Epub 2018 Feb 27. PMID: 29484647; PMCID: PMC5993571.
Huang C, Murphy GF, Akaishi S, Ogawa R. Keloids and hypertrophic scars: update and future directions. Plast Reconstr Surg Glob Open. 2013 Aug 7;1(4):e25. doi: 10.1097/GOX.0b013e31829c4597. PMID: 25289219; PMCID: PMC4173836.
Kiprono SK, Chaula BM, Masenga JE, Muchunu JW, Mavura DR, Moehrle M. Epidemiology of keloids in normally pigmented Africans and African people with albinism: population-based cross-sectional survey. Br J Dermatol. 2015 Sep;173(3):852-4. doi: 10.1111/bjd.13826. Epub 2015 Aug 13. PMID: 25833201.
Handley LJ, Manica A, Goudet J, Balloux F. Going the distance: human population genetics in a clinal world. Trends Genet. 2007; 23(9): 432-9. doi: 10.1016/j.tig.2007.07.002. Epub 2007 Jul 25. PMID: 17655965.
LeFlore IC. Misconceptions regarding elective plastic surgery in the black patient. J Natl Med Assoc. 1980 Oct;72(10):947-8. PMID: 7420436; PMCID: PMC2552534.Robles DT, Berg D. Abnormal wound healing: keloids. Clin Dermatol. 2007 Jan-Feb;25(1):26-32. doi: 10.1016/j.clindermatol.2006.09.009. PMID: 17276198.
Marneros AG, Norris JE, Olsen BR, Reichenberger E. Clinical genetics of familial keloids. Arch Dermatol. 2001 Nov;137(11):1429-34. doi: 10.1001/archderm.137.11.1429. PMID: 11708945.
Ramakrishnan KM, Thomas KP, Sundararajan CR. Study of 1,000 patients with keloids in South India. Plast Reconstr Surg. 1974 Mar;53(3):276-80. doi: 10.1097/00006534-197403000-00004. PMID: 4813760.
Rosenberg NA, Mahajan S, Ramachandran S, Zhao C, Pritchard JK, Feldman MW. Clines, clusters, and the effect of study design on the inference of human population structure. PLoS Genet. 2005; 1(6): e70. doi: 10.1371/journal.pgen.0010070. PMID: 16355252; PMCID: PMC1310579.
Chapter 3: Circulatory Dysfunction
Factor V Leiden
Approximately 2% to 15% of whites carry a specific factor V mutation (called the Leiden mutation, after the Dutch city where it was first described). (BP10 p 108)
This mutation is seen in approximately 2% to 15% of individuals of European ancestry and is present to varying degrees in other American groups, largely due to population admixture. (BP11, p. 66)
A balance between procoagulant and anticoagulant factors is necessary for normal hemostasis. Activated factor V (FV) is prothrombotic is a cofactor in the conversion of prothrombin to thrombin by factor X, leading to fibrin deposition. To maintain hemostasis and arrest thrombosis, FV is inactivated by protein C. Activated FV is also a cofactor in the inactivation of factor VIII, another protein C-mediated process.
In 1994, a group of Dutch scientists in Leiden identified a single nucleotide mutation in the gene for FV that resulted in the substitution of an arginine by a glutamine. Due to this mutation, FV is resistant to cleavage by protein C, leading to a tendency towards thrombosis (Bertina RM et al, 1994). This particular mutation is referred to factor V Leiden (FVL) and is the most common prothrombotic mutation in patients of European descent (van Mens, TE et al, 2013). Aggregated data of European populations show a carrier frequency for this mutation from 4 to 15% (Rees, DC et al, 1995; Svensson PJ et al, 1997; Zoller B et al, 1996). Frequency is higher in patients who present with venous thrombosis. Both heterozygotes and homozygotes are hypercoagulable with a 7-fold and 80-fold increased risk of thrombosis when compared to noncarriers, respectively (Rosendal FR et al, 1994).
Individuals who are not of European descent only rarely have this mutation which has been attributed to a single mutational event 24,000 to 34,000 years ago (Cox MJ et al, 1996; Zivelin A et al, 1997). Both heterozygotes and homozygotes for FVL have been identified in India which may be due to population admixture (Biswas A et al, 2008; Rees, DC et al, 1995; De Stefano V et al, 1998).
It is important to remember that patients who do not appear to have European descent may well be heterozygotes with increased thrombotic risk: studies in the United States have shown carrier rates ranging from 1.0 to 5.8% in African Americans, 2.2% in Hispanic Americans, 0.45% in Asian Americans and 1.25% in Native Americans (Mack R et al, 1998; Ridker PM et al, 1998; De Stefano V et al, 1998; Pottinger P et al, 1996). This is because African Americans, Hispanic Americans, and Native Americans all have substantial European admixture. The mean amount of European ancestry for African- and Hispanic Americans is 16% and 65—85% respectively (Bryc et al. 2015).
A carrier rate of 4 to 15% in European populations suggests that there may be an evolutionary advantage to hypercoagulability such as reduced postpartum hemorrhage and bleeding due to trauma. A selective advantage in premodern times may now be viewed as a disadvantage (evolutionary mismatch) due to a modern lifestyle that includes such as tobacco smoking, oral contraceptive pills, immobility and surgery (Rees, DC et al, 1995; Stearns and Medzhitov 2016).
In the 11th edition of Robbins, we removed the “color” language (“whites”) and emphasized the risk in non-European populations due to population admixture. The importance of this awareness is highlighted in this tragic account: https://www.npr.org/2017/12/07/568948782/black-mothers-keep-dying-after-giving-birth-shalon-irvings-story-explains-why
Bertina RM, Koeleman BP, Koster T, Rosendaal FR, Dirven RJ, de Ronde H, van der Velden PA, Reitsma PH. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994 May 5;369(6475):64-7. doi: 10.1038/369064a0. PMID: 8164741.
Biswas A, Bajaj J, Ranjan R, Meena A, Akhter MS, Yadav BK, Sharma V, Saxena R. Factor V Leiden: is it the chief contributor to activated protein C resistance in Asian-Indian patients with deep vein thrombosis? Clin Chim Acta. 2008 Jun;392(1-2):21-4. doi: 10.1016/j.cca.2008.02.018. Epub 2008 Feb 25. PMID: 18342013.
Cox MJ, Rees DC, Martinson JJ, Clegg JB. Evidence for a single origin of factor V Leiden. Br J Haematol. 1996 Mar;92(4):1022-5. doi: 10.1046/j.1365-2141.1996.4961037.x. PMID: 8616062.
Cushman M. Inherited risk factors for venous thrombosis. Hematology Am Soc Hematol Educ Program. 2005:452-7. doi: 10.1182/asheducation-2005.1.452. PMID: 16304419.
De Stefano V, Chiusolo P, Paciaroni K, Leone G. Epidemiology of factor V Leiden: clinical implications. Semin Thromb Hemost. 1998;24(4):367-79. doi: 10.1055/s-2007-996025. PMID: 9763354.
Mack R, Samaan P, Podolak I, Albanese E. Prevalence of factor V Leiden in African-Americans. Res Commun Mol Pathol Pharmacol. 1998 Mar;99(3):339-43. PMID: 9591328.
Pottinger P, Sigurdsson F, Berliner N. Detection of the factor V Leiden mutation in a nonselected black population. Blood. 1996 Mar 1;87(5):2091. PMID: 8634464.
Rees DC, Cox M, Clegg JB. World distribution of factor V Leiden. Lancet. 1995 Oct 28;346(8983):1133-4. doi: 10.1016/s0140-6736(95)91803-5. PMID: 7475606.
Ridker PM, Miletich JP, Hennekens CH, Buring JE. Ethnic distribution of factor V Leiden in 4047 men and women. Implications for venous thromboembolism screening. JAMA. 1997 Apr 23-30;277(16):1305-7. PMID: 9109469.
Rosendaal FR, Koster T, Vandenbroucke JP, Reitsma PH. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood. 1995 Mar 15;85(6):1504-8. PMID: 7888671.
Svensson PJ, Zöller B, Mattiasson I, Dahlbäck B. The factor VR506Q mutation causing APC resistance is highly prevalent amongst unselected outpatients with clinically suspected deep venous thrombosis. J Intern Med. 1997 May;241(5):379-85. doi: 10.1046/j.1365-2796.1997.124140000.x. PMID: 9183305.
Yıldız E, Türkmen FM. Factor V Leiden Mutation Frequency and Geographical Distribution in Turkish Population. J Transl Int Med. 2020 Dec 31;8(4):268-273. doi: 10.2478/jtim-2020-0040. PMID: 33511054; PMCID: PMC7805290.
Zivelin A, Griffin JH, Xu X, Pabinger I, Samama M, Conard J, Brenner B, Eldor A, Seligsohn U. A single genetic origin for a common Caucasian risk factor for venous thrombosis. Blood. 1997 Jan 15;89(2):397-402. PMID: 9002940.
Zöller B, Norlund L, Leksell H, Nilsson JE, von Schenck H, Rosén U, Jepsson JO, Dahlbäck B. High prevalence of the FVR506Q mutation causing APC resistance in a region of southern Sweden with a high incidence of venous thrombosis. Thromb Res. 1996 Sep 15;83(6):475-7. doi: 10.1016/0049-3848(96)00157-0. PMID: 8885142.
Bryc K, Durand EY, Macpherson JM, Reich D, Mountain JL. The genetic ancestry of African Americans, Latinos, and European Americans across the United States. Am J Hum Genet. 2015 Jan 8;96(1):37-53. doi: 10.1016/j.ajhg.2014.11.010. Epub 2014 Dec 18. PMID: 25529636; PMCID: PMC4289685.
Stearns SC and Medzhitov R. Evolutionary Medicine (Sunderland MA: Sinauer Asscoiates), 2016.
Chapter 4 Genetics/Pediatrics
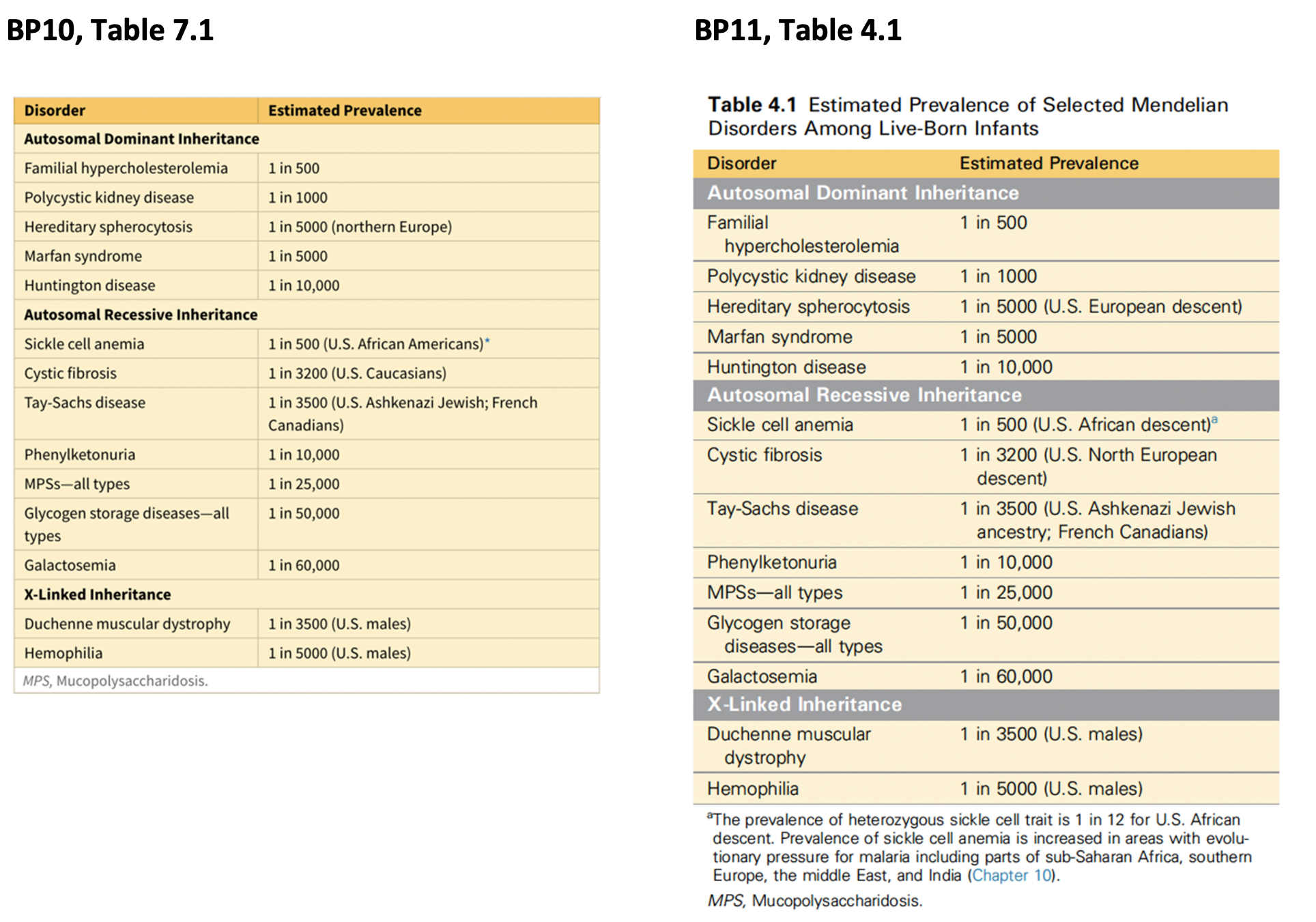
The table from the 10th edition is problematic for multiple reasons. As an educational tool it is flawed in that it gives the impression that the respective diseases (e.g., Tay-Sachs, cystic fibrosis) are seen only or primarily in particular populations, there by directing a differential diagnosis away from your clinical observations of symptoms and presentation and focusing your thoughts on the patient’s “race”. An example cited below refers to cystic fibrosis in a child of African descent who was undiagnosed until the age of 8 and only then by a radiologist who saw her chest films without knowing her race.
Another problem here is that the frequencies of all of the diseases above result from mutation/selection balance and therefore will be variable in every population surveyed. So saying that familial hypercholesterolemia is 1/500 requires that the population that this estimate was determined from be specified. Or you could say 1/500 worldwide referring to a mean estimate from all populations from which there is prevalence data for the condition. Similarly, the frequency of the sickle cell homozygote in Greeks is 0.014; while it is only 0.0016 in African Americans. Of course, terms like “Caucasian” have no legitimate place in the scientific literature and should be scrapped all together.
Since the populations are not specified (i.e., in the table, it says that PKU is seen in 1 in 10,000), this can be misleading. In the text, it specifies that the 1 in 10,000 statistic is seen in “live-born infants of European descent”.
Furthermore, the focus in the table in the 10th edition is on “race”, not geographic ancestry. Sickle cell disease, as discussed below, is a disease that evolved in parts of the world where malaria is endemic. So, first of all, one wouldn’t say African Americans because sickle cell disease is also present in Africa – it is not something that happens only to people of African descent once they arrive in the United States. Even more importantly, sickle cell disease does not affect all Africans or individuals of African descent equally: it is more prevalent in individuals from areas where malaria is endemic such as Western and Central Africa (where many enslaved peoples originated). doi:10.1016/j.gene.2010.07.008
We managed to improve the table in the 11th edition somewhat: we are now using terms related to ancestry as opposed to socially defined race. However, the populations are not clearly specified and there may be some inaccuracies in the data as presented: the original table referred to the incidence of cystic fibrosis carriers in “U.S. Caucasians”; this was simply replaced with “U.S. North European descent” as if these two populations are interchangeable. They may well be, but it depends on the original study. As you’ll read below, assessing cystic fibrosis is suboptimal in individuals who are not of European descent since the alleles assayed are predominantly European.
As always, when considering socially-defined race, one must be very careful in evaluating the source of the data. Even well-regarded sources may be full of errors and I caution you to question all “race-based” data since, as should by now be abundantly clear, human beings do not have biological races. Many of the “race” researchers are now pivoting incorrectly to the term “ethnicity.” However ethnic groups are defined as those set apart by national origin or distinctive cultural patterns (Schaefer 1996.) Thus ethnic groups such as “Hispanics” can actually differ widely in their genetic ancestry, whereas others such as Armenians may be quite uniform in their ancestry. In either case, as these are often cultural designations population admixture exists there as well (Conley AB et al, 2017).

Screening and the issues of “unique populations”: The Ashkenazi Jews
For example, germ line mutations of the familial breast/ovarian cancer gene BRCA2 are seen in approximately 10% of cases (of pancreatic cancer) arising in individuals of Ashkenazi Jewish heritage. (BP10, p 688)
Classic KS is a disorder of older men of Mediterranean, Middle Eastern, or Eastern European descent (especially Ashkenazi Jews); it is uncommon in the United States. (BP10, p 364; EP1, p 115)
The use of proactive prenatal genetic screening in high-risk populations (e.g., persons of Ashkenazi Jewish descent) has significantly reduced the incidence (Table 7.1) of certain genetic disorders such as Tay-Sachs disease. (BP10 p 246)
Tay-Sachs disease, like other lipidoses, is most common among Ashkenazi Jews, among whom the frequency of heterozygous carriers is estimated to be 1 in 30. (EP1, p 94)
In Western industrialized nations, IBD is most common among whites and, in the United States, occurs three to five times more often among eastern European (Ashkenazi) Jews. (BP10 p 621)
When I was in medical school, I remember learning about the many diseases to which Ashkenazi Jews were susceptible, but not much effort was expended in explaining why this was the case or, really, who the Ashkenazi Jews are. I now know that people of Jewish heritage are generally categorized according to origin as the Ashkenazi Jews, who originated in Central and Eastern Europe, the Sephardic Jews, who originated in Spain and Portugal and the Mizrahi Jews who originated in the Middle East and North Africa. As future practitioners, it is important for you to recognize that more than 90% of the Jewish population in the United States are Ashkenazi Jews (Brandt-Rauf, SI et al, 2006). The disease entities discussed in connection with this population are doubtless ones you will encounter during your career.
The Ashkenazim carry genetic markers that suggest they originated in the Levant and migrated to Central and Eastern Europe; the Sephardic Jews similarly originated in the Levant but migrated west to Spain and Portugal. Jewish culture rarely engaged in conversion and the mother’s religion determined the religion of the child.
There are a number of diseases that are associated with Ashkenazi heritage, including several of the diseases in this chapter: cystic fibrosis, Tay-Sachs disease, and Gaucher disease. It is not clear exactly why the allele frequencies are higher for these diseases in this population. The typical explanation would include a founder effect: a small population with deleterious mutations expanded rapidly so that the modern population has an increased incidence of these rare diseases. In the Eastern European Jewish population, ghettoization, endogamy and population contractions due to pogroms could create a founder effect. This was certainly true in the case of the WWII holocaust, as Latvia, Poland, Lithuania, Yugoslavia, Czechoslovakia, and Germany lost 89, 88, 87, 87, 83, and 83 percent of their Jewish populations respectively (Graves 2005; pg. 139).
Mutations in the BRCA1 and BRCA2 genes are also increased in incidence in this population. BRCA1 and BRCA2 are large genes and more than 2200 and 2600 pathogenic variants have been described in individuals of multiple ethnic and ancestral backgrounds (brcaexchange.org accessed 2-10-21). Certain mutations are enriched in specific populations: in the case of the Ashkenazi Jews, there are 3 particular mutations that are significantly enriched in Ashkenazi Jews. Women with BRCA1 and BRCA2 mutations are at increased risk for breast and ovarian cancer. Men with inherited BRCA2 mutations are at increased risk for prostate cancer.
A lot of genetic research has been performed in the Ashkenazi population which is a cause for some ambivalence. While identifying mutations may benefit individuals in terms of screening, prevention and treatment, there is also the potential for being viewed as a “diseased” group with multiple genetic defects (Stolberg SG, 1998 – this is a good article about the challenges a population faces).
Another issue with focus on a particular population’s “uniqueness” is that other populations with the same mutation may not be equally served or recognized. For example, Tay-Sachs is also increased in incidence in French Canadians and in the Cajun population of Louisiana (www.genome.gov/Genetic-Disorders/Tay-Sachs-Disease). While screening has reduced the incidence of Tay-Sachs in Ashkenazi Jews and in French Canadians, it is not clear that screening has had a similar effect in these populations (Kennedy J, 1990; Sugarman N, 2008; Mitchel JJ et al, 1996; Kaback MM, 2001)
In the early days of BRCA1/2 testing, there was a significant cost differential between assessing 3 founder genes vs the entire gene ($415 vs $2975) (Brandt-Rauf, SI et al, 2006). Though it is difficult to determine costs precisely in this time of pricing opacity, the current difference between a founder screen and full screen is on the order of $350 vs $950, respectively, significantly closing the gap.
One final thing to remember: the number of mutations identified in the Ashkenazi population is linked to the number of studies that have been performed in this population, not any inherent propensity for mutations (Stolberg SG, 1998). Because of endogamy and historic geographic isolation, the Ashkenazi Jews are a relatively homogeneous genetic population which facilitates identification of rare alleles. Mutations and associations are found in genomes that are searched.
Behar DM, Thomas MG, Skorecki K, Hammer MF, Bulygina E, Rosengarten D, Jones AL, Held K, Moses V, Goldstein D, Bradman N, Weale ME. Multiple origins of Ashkenazi Levites: Y chromosome evidence for both Near Eastern and European ancestries. Am J Hum Genet. 2003 Oct;73(4):768-79. doi: 10.1086/378506. Epub 2003 Sep 17. PMID: 13680527; PMCID: PMC1180600.
Graves JL. The Emperor’s New Clothes: Biological Theories of Race at the Millennium, (Rutgers University Press), 2005.
Kaback MM. Screening and prevention in Tay-Sachs disease: origins, update, and impact. Adv Genet. 2001;44:253-65. doi: 10.1016/s0065-2660(01)44084-3. PMID: 11596988.
Kennedy J. A Tragic Legacy : Why is Tay-Sachs, a rare genetic disorder, killing so many children in a tiny Cajun town? The answer seems to lie in the region’s melting-pot heritage. https://www.latimes.com/archives/la-xpm-1990-11-06-vw-4146-story.html
Mitchell JJ, Capua A, Clow C, Scriver CR. Twenty-year outcome analysis of genetic screening programs for Tay-Sachs and beta-thalassemia disease carriers in high schools. Am J Hum Genet. 1996 Oct;59(4):793-8. PMID: 8808593; PMCID: PMC1914789.
Myerowitz R. Tay-Sachs disease-causing mutations and neutral polymorphisms in the Hex A gene. Hum Mutat. 1997;9(3):195-208. doi: 10.1002/(SICI)1098-1004(1997)9:3<195::AID-HUMU1>3.0.CO;2-7. PMID: 9090523.
Neuhausen SL. Ethnic differences in cancer risk resulting from genetic variation. Cancer. 1999 Dec 1;86(11 Suppl):2575-82. doi: 10.1002/(sici)1097-0142(19991201)86:11+<2575::aid-cncr15>3.3.co;2-6. PMID: 10630184.
Petersen GM, Rotter JI, Cantor RM, Field LL, Greenwald S, Lim JS, Roy C, Schoenfeld V, Lowden JA, Kaback MM. The Tay-Sachs disease gene in North American Jewish populations: geographic variations and origin. Am J Hum Genet. 1983 Nov;35(6):1258-69. PMID: 6650504; PMCID: PMC1685967.
Risch N, Tang H, Katzenstein H, Ekstein J. Geographic distribution of disease mutations in the Ashkenazi Jewish population supports genetic drift over selection. Am J Hum Genet. 2003 Apr;72(4):812-22. doi: 10.1086/373882. Epub 2003 Feb 24. PMID: 12612865; PMCID: PMC1180346.
Sillon G, Allard P, Drury S, Rivière JB, De Bie I. The incidence and carrier frequency of Tay-Sachs disease in the French-Canadian population of Quebec based on retrospective data from 24 years, 1992-2015. J Genet Couns. 2020 Dec;29(6):1173-1185. doi: 10.1002/jgc4.1284. Epub 2020 Apr 17. PMID: 32302469.
Stolberg SG. Jewish concern grows as scientists deepen studies of Ashkenazi genes. New York Times. April 22, 1998:24.
Sugarman N. Doctors Look To Raise Tay-Sachs Awareness Among Louisiana’s Cajuns https://forward.com/culture/14042/doctors-look-to-raise-tay-sachs-awareness-among-lo-02396/
Szabo CI, King MC. Population genetics of BRCA1 and BRCA2. Am J Hum Genet. 1997 May;60(5):1013-20. PMID: 9150148; PMCID: PMC1712447.
Walsh T, Mandell JB, Norquist BM, Casadei S, Gulsuner S, Lee MK, King MC. Genetic Predisposition to Breast Cancer Due to Mutations Other Than BRCA1 and BRCA2 Founder Alleles Among Ashkenazi Jewish Women. JAMA Oncol. 2017 Dec 1;3(12):1647-1653. doi: 10.1001/jamaoncol.2017.1996. PMID: 28727877; PMCID: PMC5824270.
Newborn Screening
Newborn screening is regulated by the state and there is variability in testing modality and the selection of tests performed.
One thing to consider is how much effort/cost should be expended by society in order to identify deleterious mutations. If a condition is very rare in a particular population, the likelihood of false positives may cause more harm (e.g., additional testing, anxiety) than the “good” of diagnosing rare true positives (though perennial questions are “who is deciding what is ‘good’?” and “what is the degree of harm to undiagnosed patients?”). Cost to stretched state budgets have also moved some to argue that testing isn’t cost effective in certain populations, usually defined by socially determined race.
Another issue is how inclusive the newborn screening tests actually are. For example, for cystic fibrosis screening, the majority of screened alleles were identified in European populations (Lim RM et al, 2016). The American College of Medical Genetics and Genomics (ACMGG) recommends that screening include 23 alleles that are representative of the “pan-ethnic United States population” (Watson MS et al, 2004). Since more than 2000 CF mutations have been identified, some commercial labs have developed tests that include 90 or more alleles for screening (Lim RM, 2016). An analysis of nearly 40,000 patients with CF identified 159 variants had an allele frequency of ≥0.01%, the threshold recommended by the ACMGG. Of those variants, 127 caused disease. However, 95% of the 40,000 patients in this database were … of European descent. One consequence of this is that a negative CF screening test in a non-European patient is not as conclusive as a negative test in a patient of European descent.
This image from Lim and colleagues (Lim RM et al, 2016) shows predicted carrier detection rates for 5 common screening platforms (Counsyl, Recombine, ACMG, Integrated genetics and CFTR2) for group 1 (variants likely to cause CF) in a variety of populations based on targeted mutation screening panels (the methodology is a bit detailed for this document, but worth reading about in the original article). The data demonstrate that the ACMG test (which provides practice guidelines in the US), is particularly bad at identifying potential disease-causing mutations in non-European populations, particularly in patients of East Asian descent.
There has not been much research into CF alleles in non-Europeans. An extensive review of the literature in 2016 by Stewart and Pepper of reported cases of CF with mutational analysis in Africa found reports from only 12 of the 54 African countries (Stewart C et al, 2016). 79 different mutations were identified, of which 21 were unique to Africa.

Dr. Lainie Ross is a pediatric bioethicist from the University of Chicago, who has devoted a lot of thought to this issue in the context of screening for cystic fibrosis (Ross LR, 2008). Some of the issues she raises in her article “Newborn Screening for Cystic Fibrosis: A Lesson in Public
Health Disparities” have been solved by the decreased cost in testing due to advances in technology. Affordable and more comprehensive testing has become the norm, which reduces racial disparities in CF screening. However, the questions are still relevant regarding tests that may be developed in the future: Who needs it? Who gets it? Who decides? Who pays for it? This article also addresses the ethics of newborn screening such as the decision of parents who may or may not want to know their own carrier status and disclosure of a condition to an infant who cannot give consent to gain this knowledge. Such considerations will be something for you to remember during your pediatrics rotations next year.
One final point that Dr. Ross shared with me in an email: in the consideration of public health, it is extremely important to pick up individuals who are not of European descent by screening because, otherwise, there may be a significant delay in diagnosis: when a 2-month-old child of African descent presents in the ED with failure to thrive, physicians may first worry about neglect, not cystic fibrosis; for a 2-month-old child of European descent presenting with failure to thrive, it is more likely that the concern will first be cystic fibrosis, then neglect. Universal newborn screening can help reduce health disparities (Brosco JP et al, 2015).
This is why it is critical that you look at your patients with open minds and evaluate all of the data. Do not discount a diagnosis simply because “it is uncommon in this race.”
Bobadilla JL, Macek M Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations--correlation with incidence data and application to screening. Hum Mutat. 2002 Jun;19(6):575-606. doi: 10.1002/humu.10041. PMID: 12007216.
Brosco JP, Grosse SD, Ross LF. Universal state newborn screening programs can reduce health disparities. JAMA Pediatr. 2015 Jan;169(1):7-8. doi: 10.1001/jamapediatrics.2014.2465. PMID: 25402722; PMCID: PMC4528613.
Grody WW, Cutting GR, Klinger KW, Richards CS, Watson MS, Desnick RJ; Subcommittee on Cystic Fibrosis Screening, Accreditation of Genetic Services Committee, ACMG. American College of Medical Genetics. Laboratory standards and guidelines for population-based cystic fibrosis carrier screening. Genet Med. 2001 Mar-Apr;3(2):149-54. doi: 10.1097/00125817-200103000-00010. PMID: 11280952.
Lim RM, Silver AJ, Silver MJ, Borroto C, Spurrier B, Petrossian TC, Larson JL, Silver LM. Targeted mutation screening panels expose systematic population bias in detection of cystic fibrosis risk. Genet Med. 2016 Feb;18(2):174-9. doi: 10.1038/gim.2015.52. Epub 2015 Apr 16. PMID: 25880441.
Ross LF. Newborn screening for cystic fibrosis: a lesson in public health disparities. J Pediatr. 2008 Sep;153(3):308-13. doi: 10.1016/j.jpeds.2008.04.061. PMID: 18718257; PMCID: PMC2569148.
Sosnay PR, Siklosi KR, Van Goor F, Kaniecki K, Yu H, Sharma N, Ramalho AS, Amaral MD, Dorfman R, Zielenski J, Masica DL, Karchin R, Millen L, Thomas PJ, Patrinos GP, Corey M, Lewis MH, Rommens JM, Castellani C, Penland CM, Cutting GR. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet. 2013 Oct;45(10):1160-7. doi: 10.1038/ng.2745. Epub 2013 Aug 25. PMID: 23974870; PMCID: PMC3874936.
Stewart C, Pepper MS. Cystic fibrosis on the African continent. Genet Med. 2016 Jul;18(7):653-62. doi: 10.1038/gim.2015.157. Epub 2015 Dec 10. Erratum in: Genet Med. 2016 Apr;18(4):418. PMID: 26656651.
Watson MS, Cutting GR, Desnick RJ, Driscoll DA, Klinger K, Mennuti M, Palomaki GE, Popovich BW, Pratt VM, Rohlfs EM, Strom CM, Richards CS, Witt DR, Grody WW. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med. 2004 Sep-Oct;6(5):387-91. doi: 10.1097/01.gim.0000139506.11694.7c. Erratum in: Genet Med. 2004 Nov-Dec;6(6):548. Erratum in: Genet Med. 2005 Apr;7(4):286. PMID: 15371902; PMCID: PMC3110945.
Cystic fibrosis
The carrier frequency [of cystic fibrosis] in the United States is 1 in 20 among Caucasians but significantly lower among African Americans, Asians, and Hispanics. (BP10, p 251)
Cystic fibrosis is due to mutations in the CFTR gene which encodes a calcium channel that additionally regulates multiple ion channels (e.g., epithelial sodium channel, gap junction channels) and the transport of bicarbonate. In the lung, decreased chloride secretion and increased sodium and water resorption results in thickened bronchial secretions, an ideal medium for the bacterium Pseudomonas aeruginosa. Up until the 1960’s, CF was primarily a pediatric disease since most patients died at about six years of age. Most patients in the United States now live into their 40’s due to a combination of improved nutrition and physical therapies and better medical therapies aimed at airway clearance and infections. CFTR-modulating drugs are a new class of drugs that may also have an impact on survival.
The most frequent cystic fibrosis mutation, F508del has its greatest prevalence in northwest Europe and declining incidence as one moves southeast. It appears that this mutation arose in the Bronze age in the Bell Beaker folk who slowly migrated west to east, which would account for the geographic distribution (Philip F et al, 2018).
No evolutionary advantage has been linked to this mutation, though there have been some proposals: both cholera and typhoid fever have been considered possible factors. Salmonella typhi binds to the CFTR protein in the intestines; however, it cannot bind as effectively to mutant CFTR, reducing infectivity (by contrast, in the lung this same defect causes increased morbidity due to decreased clearance of pulmonary Pseudomonas aeruginosa) (Pier G et al, 1998). Cholera infection, which results in massive secretory diarrhea, was theorized to be less morbid in CF heterozygotes since, presumably, they would lose less water; however, cholera didn’t affect Northern Europe until the 19th century. For both typhoid fever and cholera, the mortality rate is too low to explain maintenance of the allele.
About 1 in 20 individuals from European populations and the Ashkenazi Jewish population are carriers for CF. In addition, CF is increasingly recognized in non-European populations, including South and East Asia, Africa, and Latin America; overall prevalence in these regions is low (Yamashiro Y et al, 1997; Stewart C and Pepper MS, 2015; Guo X et al; 2018). One challenge is that since CF has long been considered a disease in people of European descent, research hasn’t focused on other populations. Another issue is that lower- and middle-income countries often lack resources for screening.
Of course, cystic fibrosis DOES affect patients who are not of European descent. Furthermore, in the United States, due to significant admixture with European populations, a patient’s phenotype is only a poor measure of his/her/their genotype. Not including this disease in your differential diagnosis can lead to delay in diagnosis as in this story: My childhood friend Lela wasn’t diagnosed with cystic fibrosis until she was 8 years old. Over the years, her doctors had described her as a “2-year-old black female with fever and cough...4-year-old black girl with another pneumonia. Lela is back.” Had she been a white child, or had no visible “race” at all, she would probably have gotten the correct diagnosis and treatment much earlier. Only when she was 8 did a radiologist, who had never seen her face to face, notice her chest radiograph and ask, “Who’s the kid with CF?” (Garcia RS, 2004).
Early diagnosis is critical: a patient with CF in South Africa has a life expectancy of 20.5 years, in contrast to the 40+ years in the United States (Stewart CS and Pepper MS, 2016).
Without the correct diagnosis, the correct treatment cannot be given.
With an incidence of 1 in 2500 live births in the United States, CF is the most common life-limiting genetic disease that affects individuals of European descent. The carrier frequency in the United States is 1 in 20 among individuals of European descent but significantly lower among individuals of other ancestral origins. (BP11, p. 89)
In the 11th edition, we have removed the term “Caucasian” and replaced it with terms related to ancestry since this is a single gene disorder.
Farrell PM. The prevalence of cystic fibrosis in the European Union. J Cyst Fibros. 2008 Sep;7(5):450-3. doi: 10.1016/j.jcf.2008.03.007. Epub 2008 Apr 28. PMID: 18442953.
Farrell P, Férec C, Macek M, et al. Estimating the age of p.(Phe508del) with family studies of geographically distinct European populations and the early spread of cystic fibrosis. Eur J Hum Genet. 2018 Dec;26(12):1832-1839. doi: 10.1038/s41431-018-0234-z. Epub 2018 Aug 8. PMID: 30089827; PMCID: PMC6244163.
Garcia RS. The misuse of race in medical diagnosis. Pediatrics. 2004 May;113(5):1394-5. doi: 10.1542/peds.113.5.1394. PMID: 15121958.
Goodman BE, Percy WH. CFTR in cystic fibrosis and cholera: from membrane transport to clinical practice. Adv Physiol Educ. 2005 Jun;29(2):75-82. doi: 10.1152/advan.00035.2004. PMID: 15905150.
Guo X, Liu K, Liu Y, Situ Y, Tian X, Xu KF, Zhang X. Clinical and genetic characteristics of cystic fibrosis in CHINESE patients: a systemic review of reported cases. Orphanet J Rare Dis. 2018 Dec 17;13(1):224. doi: 10.1186/s13023-018-0968-2. PMID: 30558651; PMCID: PMC6296146.
Hamosh A, FitzSimmons SC, Macek M Jr, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifestations of cystic fibrosis in black and white patients. J Pediatr. 1998 Feb;132(2):255-9. doi: 10.1016/s0022-3476(98)70441-x. PMID: 9506637.
Poolman EM, Galvani AP. Evaluating candidate agents of selective pressure for cystic fibrosis. J R Soc Interface. 2007 Feb 22;4(12):91-8. doi: 10.1098/rsif.2006.0154. PMID: 17015291; PMCID: PMC2358959.
Stewart C, Pepper MS. Cystic fibrosis on the African continent. Genet Med. 2016 Jul;18(7):653-62. doi: 10.1038/gim.2015.157. Epub 2015 Dec 10. Erratum in: Genet Med. 2016 Apr;18(4):418. PMID: 26656651.
Yamashiro Y, Shimizu T, Oguchi S, Shioya T, Nagata S, Ohtsuka Y. The estimated incidence of cystic fibrosis in Japan. J Pediatr Gastroenterol Nutr. 1997 May;24(5):544-7. doi: 10.1097/00005176-199705000-00010. PMID: 9161949.
Tay-Sachs disease
Tay-Sachs disease was initially described in 1881 by Warren Tay based on yellowish degeneration of the fundi of a child with a neurodegenerative disorder (Kaback MM and Desnick RJ, 2001). Five years later, Bernard Sachs described several additional patients who, he noted, were of Eastern European Jewish (Ashkenazi) descent. Tay-Sachs disease is relentlessly progressive disease that typically results in death by the age of four years. There is no cure, though some treatments can help with symptoms.
By the 1930’s, the autosomal recessive pattern of inheritance had been noted and the association with Eastern European Jewish heritage solidified. In 1969, the defect in hexosaminidase A was identified. By 1979, a rapid screening test of hexosaminidase A activity was possible and could identify carriers, thereby enabling prospective parents to determine their degree of risk for having a child with Tay-Sachs disease. When antenatal diagnosis became possible, parents had the choice of continuing or aborting the pregnancy. The incidence of Tay-Sachs disease has dropped significantly in the Ashkenazi Jewish population as well as in French Canadians, in large measure due to screening (Kaback MM, 2001; Mitchel JJ et al, 1996).
In the Ashkenazi Jewish population, there are 3 mutations that account for about 96% of cases of Tay-Sachs disease: two of the mutations are for the infantile form and the third is an adult-onset type (Paw BH et al, 1990). Among the French-Canadians, there are two mutations, both of fairly recent origin (after the British conquest of Quebec in 1759) (De Braekeleer M et al, 1992).
In the 11th edition of Robbins, we include information about how the evolutionary processes that could have contributed to the increased disease prevalence in the Ashkenazim and further described this population in greater detail (BP11, p 95):
Due to founder effects, Tay-Sachs disease, similar to other lipid storage disorders, has an increased prevalence among individuals of Ashkenazi Jewish ancestry, among whom the frequency of heterozygous carriers is estimated to be 1 in 30. The Ashkenazim originated in Eastern and Central Europe and constitute more than 90% of the Jewish population in the United States.
Aronsont SM. Early epidemiologic studies of Tay-Sachs disease. Adv Genet. 2001;44:25-31. doi: 10.1016/s0065-2660(01)44067-3. PMID: 11596987.
De Braekeleer M, Hechtman P, Andermann E, Kaplan F. The French Canadian Tay-Sachs disease deletion mutation: identification of probable founders. Hum Genet. 1992 Apr;89(1):83-7. doi: 10.1007/BF00207048. PMID: 1577470.
Kaback MM. Screening and prevention in Tay-Sachs disease: origins, update, and impact. Adv Genet. 2001;44:253-65. doi: 10.1016/s0065-2660(01)44084-3. PMID: 11596988.
Kaback MM, Desnick RJ. Tay-Sachs disease: from clinical description to molecular defect. Adv Genet. 2001;44:1-9. doi: 10.1016/s0065-2660(01)44065-x. PMID: 11596975.
Mitchell JJ, Capua A, Clow C, Scriver CR. Twenty-year outcome analysis of genetic screening programs for Tay-Sachs and beta-thalassemia disease carriers in high schools. Am J Hum Genet. 1996 Oct;59(4):793-8. PMID: 8808593; PMCID: PMC1914789.
Paw BH, Tieu PT, Kaback MM, Lim J, Neufeld EF. Frequency of three Hex A mutant alleles among Jewish and non-Jewish carriers identified in a Tay-Sachs screening program. Am J Hum Genet. 1990 Oct;47(4):698-705. PMID: 2220809; PMCID: PMC1683802.
Sillon G, Allard P, Drury S, Rivière JB, De Bie I. The incidence and carrier frequency of Tay-Sachs disease in the French-Canadian population of Quebec based on retrospective data from 24 years, 1992-2015. J Genet Couns. 2020 Dec;29(6):1173-1185. doi: 10.1002/jgc4.1284. Epub 2020 Apr 17. PMID: 32302469.
Phenylketonuria
The most common form, referred to as classic phenylketonuria, is common in persons of Scandinavian descent and is distinctly uncommon in African-American and Jewish populations. (BP10 p 254;
The most common (classic) form is relatively common in persons of Scandinavian descent and uncommon in Jewish populations and in persons of African descent. (EP1, p 94)
Phenylketonuria (PKU) is the most frequent genetic disease of amino acid metabolism and is an example of how screening can have a transformational effect on a patient’s health. Infants who are homozygous for mutations that reduce or eliminate the activity of the enzyme phenylalanine hydroxylase (PAH) experience hyperphenyalaninemia which leads to severe intellectual disability and seizures as well as other symptoms. By adopting a phenylalanine-poor diet early in life, however, affected individuals can greatly reduce the majority of the symptoms and have a typical lifespan. In contrast to the information in Basic Pathology and Essential Pathology, it is now recommended that dietary restrictions be maintained for life.
Although PKU is often described as a disease of individuals of European descent, it is now known that there is wide variation in the global distribution (Hillert A et al, 2020):

Previously, the longstanding bias that PKU did not occur in non-European individuals had an impact on early thinking about newborn screening: In the initial field trial of the Guthrie PKU test in the 1960’s, to increase cost effectiveness, nonwhite infants were excluded from testing (Ross LF et al, 2015; Paul DB and Brosco JP, 2014). Race-based screening was not ultimately adopted for PKU. It is worth remembering, however, that as recently as 2015, the American College of Obstetricians and Gynecologists only recommended prenatal screening for certain hemoglobinopathies in a subset of racial/ethnic groups (Ross LF et al, 2015; ACOG, 2007). Current recommendations are for ALL pregnant women to undergo prenatal screening, in recognition of the fact that race does not correlate with genotype (www.acog.org/womens-health/faqs/carrier-screening-for-hemoglobinopathies). For PKU, this distinction is particularly important since the consequences of missing a diagnosis for an easily treatable disease are so consequential.
In the 11th edition, we made the discussion a bit more general. However, this is still problematic. For example, the Finns (Europeans) have a very low rate of PKU. UpToDate makes the point that prevalence varies widely across different regions. If we continue to emphasize the prevalence in European populations, we set ourselves up to MISS this disease in other groups! Also, note that in the new table, the 1 in 10,000 incidence is not qualified by population. According to the text, this is for European descent; therefore, the table is misleading.
Updated version in the 11th edition of Basic Pathology: It [PKU] affects 1 in 10,000 live-born infants of European descent, and there are several variants of this disease. The most common form is referred to as classic phenylketonuria; its incidence is higher in European populations and less common in individuals from other geographic regions. (BP11 p 92)
ACOG Committee on Obstetrics. ACOG practice bulletin no. 78: hemoglobinopathies in pregnancy. Obstet Gynecol 2007;109:229-37.
Hillert A, Anikster Y, Belanger-Quintana A et al. The Genetic Landscape and Epidemiology of Phenylketonuria. Am J Hum Genet. 2020 Aug 6;107(2):234-250. doi: 10.1016/j.ajhg.2020.06.006. Epub 2020 Jul 14. PMID: 32668217; PMCID: PMC7413859.
Hofman KJ, Steel G, Kazazian HH, Valle D. Phenylketonuria in U.S. blacks: molecular analysis of the phenylalanine hydroxylase gene. Am J Hum Genet. 1991 Apr;48(4):791-8. PMID: 2014802; PMCID: PMC1682942.
Paul DB, Brosco JP. The PKU paradox. Baltimore, MD: JHU Press; 2014.
Ross LF, Paul DB, Brosco JP. 50 Years Ago in The Journal of Pediatrics: Phenylketonuria in a Negro Infant. J Pediatr. 2015 Aug;167(2):304. doi: 10.1016/j.jpeds.2015.01.048. PMID: 26210837.
Wang T, Okano Y, Eisensmith RC, Harvey ML, Lo WH, Huang SZ, Zeng YT, Yuan LF, Furuyama JI, Oura T, et al. Founder effect of a prevalent phenylketonuria mutation in the Oriental population. Proc Natl Acad Sci U S A. 1991 Mar 15;88(6):2146-50. doi: 10.1073/pnas.88.6.2146. PMID: 2006152; PMCID: PMC51186.
Chapter Five: Immunopathology
Systemic lupus erythematosus
The prevalence of the disease is 2- to 3-fold higher in blacks and Hispanics than in whites. (BP10 p 150).
The prevalence and severity of the disease are higher in African-Americans and Latin-Americans than in European-Americans in the United States (BP11 p 153)
Lupus is a severe, chronic multisystem autoimmune disease that, because of its protean manifestations, can be challenging to diagnose. Affected organs include the kidneys, skin, bone marrow, gastrointestinal tract, and central nervous system. Part of the challenge in diagnosis is that the criteria continue to evolve and involve both clinical findings and laboratory tests (Aringer M et al, 2019). The incidence of SLE is higher in non-European populations and is associated with earlier onset and increased morbidity. Mortality rates are also higher in these patients. Some of this disparity is likely genetic and the exact social/environmental contributions are not clear.
As with most autoimmune diseases, women are affected to a greater degree than men.
Multiple studies have consistently demonstrated racial and ethnic differences in lupus incidence and outcome. One recent example is a meta-analysis from the University of Michigan of 4 registries in the United States (Izmirly PM et al, 2021). Rates are per 100,000 individuals (see table at right).

SLE has a strong genetic component with a high concordance rate in monozygotic twins and first-degree family members (Deapen D et al, 1992; Alarcón-Segovia D et al, 2005; Kuo CF et al, 2015). There is likely a great degree of heterogeneity in the molecular pathogenesis of this disease. Moreover, environmental and epigenetic factors play a substantial and complicated role in pathogenesis. Although these factors remain poorly defined, candidate factors include smoking, exposure to crystalline silica (found in some cleaning products), exposure to ultraviolet light, oral contraceptives, and infections, especially by trypanosomes, mycobacteria and Epstein-Barr virus (UpToDate; Parks CG et al, 2017).
Regarding epigenetics, a recent paper has identified differential methylation of specific CpG islands that correlate with disease severity and ethnicity (Lanata C et al, 2019). As you’ll recall, methylation of CpG islands abrogates gene expression. One important point that the researchers make is this: although there can be “ethnicity-associated” CpG island methylation, we can’t distinguish between methylation associated with population ancestry and methylation due to environmental exposures. Just because something looks “ethnicity-associated” doesn’t mean it’s genetic. Community groups share cultural heritage, customs, foods, activities and geopolitical factors - any of which could be associated with an environmental epigenetic impact.
A useful tool to determine the impact of environmental versus genetic factors is to compare disease incidence in the country of ancestry to incidence in the United States. Although earlier reports suggested that the incidence of SLE in Africa was low, more recent studies have indicated that the disease is prevalent on the continent (Tiffin N et al, 2014; Essouma M et al, 2020). It is likely that the true incidence was underreported in earlier studies due to 1) diagnostic delay; 2) lack of disease recognition at point of care and in patient populations; 3) lack of specialty physicians; and 4) limited availability of diagnostic tools.
Morbidity and mortality are higher in non-European patients with SLE. Lupus nephritis, a major factor in the morbidity and mortality of lupus, is more prevalent in African Americans, Hispanic Americans, Chinese and a subset of other Asians (e.g., Thai, Korean) (Lau CS et al, 2006). Cardiac and hematologic findings (e.g., cytopenias) findings are also more common in African Americans (Uribe AG et al, 2004) which may contribute to increased mortality.
Other factors clearly link with morbidity and mortality, including availability of private health insurance, educational level and poverty (Uribe AG et al, 2004). Patients of lower socioeconomic status may experience delay in diagnosis, lack of access to specialized care and appropriate treatment and may face challenges in paying for immunosuppressant medication.
I believe that labeling socially defined races as having a higher likelihood (“risk”) of certain diseases is sloppy science. We are using socially defined race as a proxy for social determinants of health and (perhaps) some genetic predisposition. This genetic predisposition is associated with populations, not socially defined races.
Furthermore, it is often said that race-based medicine is useful in developing a differential diagnosis. However, any person can present with any disease; making race-based assumptions can lead to delay in diagnosis and patient harm. Also, when we are discussing diseases that are very rare (e.g., lupus), the difference between 85 patients in 100,000 (European American women), 121 patients in 100,000 (“Hispanic” women) and 231 patients in 100,000 (African American women) is of minimal clinical significance. You could not ignore signs/symptoms/laboratory values suggestive of lupus in a European American woman simply because of her socially defined race.
Despite our efforts, Dr. Graves and I were unable to remove this race-based association from the 11th edition.
Alarcón-Segovia D, Alarcón-Riquelme ME, Cardiel MH, Caeiro F, Massardo L, Villa AR, Pons-Estel BA; Grupo Latinoamericano de Estudio del Lupus Eritematoso (GLADEL). Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum. 2005 Apr;52(4):1138-47. doi: 10.1002/art.20999. PMID: 15818688.
Aringer M, et al. 2019 European League Against Rheumatism/American College of Rheumatology Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019 Sep;71(9):1400-1412. doi: 10.1002/art.40930. Epub 2019 Aug 6. PMID: 31385462; PMCID: PMC6827566.
Deapen D, Escalante A, Weinrib L, Horwitz D, Bachman B, Roy-Burman P, Walker A, Mack TM. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum. 1992 Mar;35(3):311-8. doi: 10.1002/art.1780350310. PMID: 1536669.
Essouma M, Nkeck JR, Endomba FT, Bigna JJ, Singwe-Ngandeu M, Hachulla E. Systemic lupus erythematosus in Native sub-Saharan Africans: A systematic review and meta-analysis. J Autoimmun. 2020 Jan;106:102348. doi: 10.1016/j.jaut.2019.102348. Epub 2019 Oct 23. PMID: 31668352.
Falasinnu T, Chaichian Y, Palaniappan L, Simard JF. Unraveling Race, Socioeconomic Factors, and Geographical Context in the Heterogeneity of Lupus Mortality in the United States. ACR Open Rheumatol. 2019 Apr 29;1(3):164-172. doi: 10.1002/acr2.1024. PMID: 31777791; PMCID: PMC6858029.
Hodkinson B, Mapiye D, Jayne D, Kalla A, Tiffin N, Okpechi I. The African Lupus Genetics Network (ALUGEN) registry: standardized, prospective follow-up studies in African patients with systemic lupus erythematosus. Lupus. 2016 Mar;25(3):325-30. doi: 10.1177/0961203315606984. Epub 2015 Sep 24. PMID: 26405020.
Izmirly PM, Parton H, Wang L, McCune WJ, Lim SS, Drenkard C, Ferucci ED, Dall'Era M, Gordon C, Helmick CG, Somers EC. Prevalence of Systemic Lupus Erythematosus in the United States: Estimates from a Meta-Analysis of the Centers for Disease Control and Prevention National Lupus Registries. Arthritis Rheumatol. 2021 Jan 20. doi: 10.1002/art.41632. Epub ahead of print. PMID: 33474834.
Kuo CF, Grainge MJ, Valdes AM, See LC, Luo SF, Yu KH, Zhang W, Doherty M. Familial Aggregation of Systemic Lupus Erythematosus and Coaggregation of Autoimmune Diseases in Affected Families. JAMA Intern Med. 2015 Sep;175(9):1518-26. doi: 10.1001/jamainternmed.2015.3528. PMID: 26193127.
Lanata CM, Paranjpe I, Nititham J, Taylor KE, Gianfrancesco M, Paranjpe M, Andrews S, Chung SA, Rhead B, Barcellos LF, Trupin L, Katz P, Dall'Era M, Yazdany J, Sirota M, Criswell LA. A phenotypic and genomics approach in a multi-ethnic cohort to subtype systemic lupus erythematosus. Nat Commun. 2019 Aug 29;10(1):3902. doi: 10.1038/s41467-019-11845-y. Erratum in: Nat Commun. 2020 Feb 27;11(1):1164. PMID: 31467281; PMCID: PMC6715644.
Lau CS, Yin G, Mok MY. Ethnic and geographical differences in systemic lupus erythematosus: an overview. Lupus. 2006;15(11):715-9. doi: 10.1177/0961203306072311. PMID: 17153840.
Lewis MJ, Jawad AS. The effect of ethnicity and genetic ancestry on the epidemiology, clinical features and outcome of systemic lupus erythematosus. Rheumatology (Oxford). 2017 Apr 1;56(suppl_1):i67-i77. doi: 10.1093/rheumatology/kew399. PMID: 27940583.
Parks CG, de Souza Espindola Santos A, Barbhaiya M, Costenbader KH. Understanding the role of environmental factors in the development of systemic lupus erythematosus. Best Pract Res Clin Rheumatol. 2017 Jun;31(3):306-320. doi: 10.1016/j.berh.2017.09.005. Epub 2017 Oct 21. PMID: 29224673; PMCID: PMC5729939.
Tiffin N, Hodkinson B, Okpechi I. Lupus in Africa: can we dispel the myths and face the challenges? Lupus. 2014;23(1):102-11. doi: 10.1177/0961203313509296. Epub 2013 Oct 30. PMID: 24174511.
Uribe AG, McGwin G Jr, Reveille JD, Alarcón GS. What have we learned from a 10-year experience with the LUMINA (Lupus in Minorities; Nature vs. nurture) cohort? Where are we heading? Autoimmun Rev. 2004 Jun;3(4):321-9. doi: 10.1016/j.autrev.2003.11.005. PMID: 15246029.
Yen EY, Shaheen M, Woo JMP, Mercer N, Li N, McCurdy DK, Karlamangla A, Singh RR. 46-Year Trends in Systemic Lupus Erythematosus Mortality in the United States, 1968 to 2013: A Nationwide Population-Based Study. Ann Intern Med. 2017 Dec 5;167(11):777-785. doi: 10.7326/M17-0102. Epub 2017 Oct 31. PMID: 29086801; PMCID: PMC6188647.
Severe Combined Immunodeficiency
The overall prevalence of the disease [severe combined immunodeficiency] is approximately 1 in 65,000 to 1 in 100,000, but it is 20 to 30 times more frequent in some Native American populations. (BP10 p 168)
The overall prevalence of the disease is approximately 1 in 65,000 to 1 in 100,000, but it is 20 to 30 times more frequent in Navajo and Apache American Indian populations (BP11, p. 166).
Severe combined immunodeficiency (SCID) is an umbrella term for a number of phenotypically and genetically diverse diseases that share the common feature of severe B- and T-cell immunodeficiency. Athabascan-type SCID (SCIDA) is a T-B-NK+ variant that is characterized by a near absence of circulating B and T cell but normal levels of correctly functioning natural killer cells. While this autosomal recessive disease is extremely rare in the general population (about 1:1,000,000 live births), the incidence in two Athabascan-speaking American Indian tribes, the Navajo and Jicarilla Apache, is extremely high at 1:2000 live births (Kwan A et al, 2015; Murphy S et al, 1980). In these populations, the disease is associated with oral and genital ulcerations (Kwong PC et al, 1999) as well as the usual findings of early onset, recurrent infections and failure to thrive.
V(D)J recombination in the antigen recognition receptors of B and T cells is initiated by the RAG1/2 complex. Mutations in the genes that encode RAG1 and RAG2 are one etiology of SCID. Similarly, in SCIDA, a nonsense mutation the DCLRE1C gene abrogates function of a protein (Artemis) involved in V(D)J recombination and DNA repair (Li L et al, 2002).
The high incidence of SCIDA in these populations is due to a founder mutation due to a population bottleneck (Li L et al, 2002). From 1863-1868, the Navajo population was interned at Fort Defiance in Arizona from which multiple treks of 250 – 400 miles (depending on the route), including “The Long Walk”, were made to Fort Sumner in New Mexico territory (Kwan A et al, 2015). Approximately 10,000 Navajo began the journey, but many died en route or at the New Mexican camp (https://americanindian.si.edu/nk360/navajo/long-walk/long-walk.cshtml). It is thought that about 8,000 survived. From that small population, the Navajo now number about 330,000. The Jicarilla Apache declined to a population of approximately 600 in the early 20th century and now number about 3,300. For more information on Native American history in the United States, see https://www.nlm.nih.gov/nativevoices/timeline/index.html.
Early detection of SCID is critical to prevent life-threatening infections that typically occur during the first six months of life. With identification of the causative mutation of SCIDA, newborn screening has been implemented in populations at high risk (Kwan A et al, 2015). Hematopoietic stem cell transplantation is the definitive therapy.
One more comment: “Athabascan-type” SCID refers to the variant seen in this population of Native Americans. There is some stigmatization in using such terms: think “the Chinese virus” or “Navajo neuropathy”. Diseases are rarely limited to a single population and, in fact, both SCIDA and the so-called “Navajo neuropathy” have been diagnosed in non-native populations. Words matter (Begay RL et al, 2020).
In the 11th edition, we specified the populations that are associated with hereditary SCID (i.e., Navajo and Apache). This underscores the fact that this heritable disease affects populations (Navajo, Apache) not socially defined races (“American Indian”).
Begay RL, Garrison NA, Sage F, Bauer M, Knoki-Wilson U, Begay DH, Becenti-Pigman B, Claw KG. Weaving the Strands of Life (Iiná Bitł'ool): History of Genetic Research Involving Navajo People. Hum Biol. 2020 Jul 9;91(3):189-208. doi: 10.13110/humanbiology.91.3.04. PMID: 32549035.
Jones JF, Ritenbaugh CK, Spence MA, Hayward A. Severe combined immunodeficiency among the Navajo. I. Characterization of phenotypes, epidemiology, and population genetics. Hum Biol. 1991 Oct;63(5):669-82. PMID: 1916741.
Kwan A et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014 Aug 20;312(7):729-38. doi: 10.1001/jama.2014.9132.
Kwan A, Hu D, Song M, Gomes H, Brown DR, Bourque T, Gonzalez-Espinosa D, Lin Z, Cowan MJ, Puck JM. Successful newborn screening for SCID in the Navajo Nation. Clin Immunol. 2015 May;158(1):29-34. doi: 10.1016/j.clim.2015.02.015. Epub 2015 Mar 8. PMID: 25762520; PMCID: PMC4420660.
Kwong PC, O'Marcaigh AS, Howard R, Cowan MJ, Frieden IJ. Oral and genital ulceration: a unique presentation of immunodeficiency in Athabascan-speaking American Indian children with severe combined immunodeficiency. Arch Dermatol. 1999 Aug;135(8):927-31. doi: 10.1001/archderm.135.8.927. PMID: 10456341.
Li L, Moshous D, Zhou Y, Wang J, Xie G, Salido E, Hu D, de Villartay JP, Cowan MJ. A founder mutation in Artemis, an SNM1-like protein, causes SCID in Athabascan-speaking Native Americans. J Immunol. 2002 Jun 15;168(12):6323-9. doi: 10.4049/jimmunol.168.12.6323. PMID: 12055248.
Murphy S, Hayward A, Troup G, Devor EJ, Coons T. Gene enrichment in an American Indian population: an excess of severe combined immunodeficiency disease. Lancet. 1980 Sep 6;2(8193):502-5. doi: 10.1016/s0140-6736(80)91833-4. PMID: 6105560.
Cardiac amyloidosis
“The mutated form of the TTR gene that leads to cardiac amyloidosis is carried by approximately 4% of the black population in the United States, and cardiomyopathy has been identified in both homozygous and heterozygous patients.” (BP10, p 433)
Four percent of African Americans carry a specific mutation of transthyretin that increases the risk of cardiac amyloidosis in that population over fourfold. (BP10, p 185)
A variant of the TTR gene that leads to cardiac amyloidosis is carried by approximately 4% of the African American population in the United States; restrictive cardiomyopathy has been identified in both homozygous and heterozygous patients. The origin of the allele in this population appears to be in West African regions where much of the African American population of the United States originated. (BP11, p 179)
Four percent of African Americans carry a specific mutation of transthyretin that increases the risk of cardiac amyloidosis in that population over fourfold. (BP11, p. 338)
Transthyretin is a homotetramer composed of four 127-amino acid peptides that are rich in beta sheets. Mutations that destabilize the tetramer allow the monomers to aggregate as amyloid in peripheral nerves and the heart which can lead to polyneuropathy and cardiac arrhythmias and heart failure, respectively. The latter entity, called hATTR-associated cardiomyopathy (hATTR-CM), mimics hypertensive, hypertrophic cardiac disease and because of its rarity in the general population is not often considered diagnostically (Maurer MS, 2018).
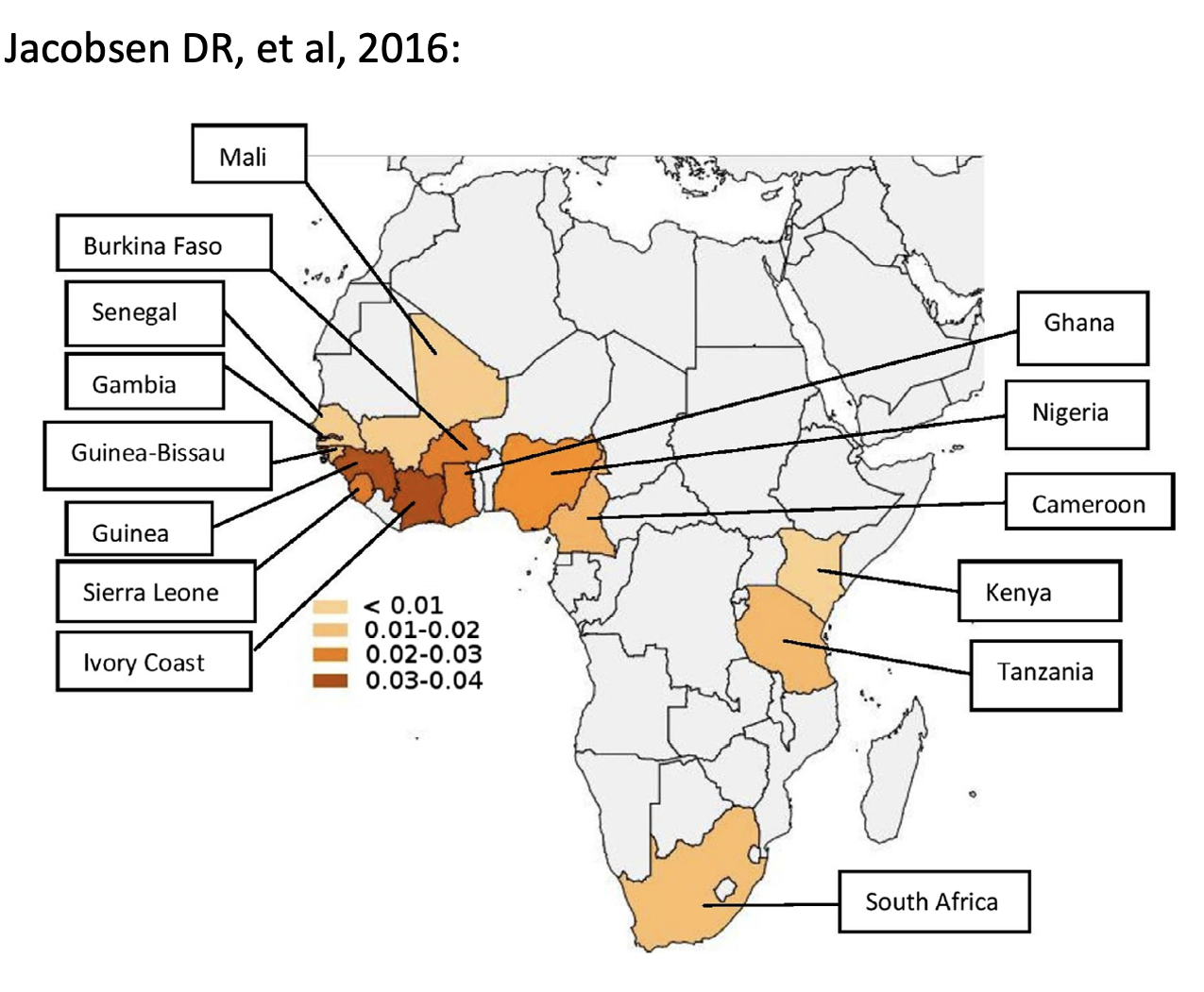
The most common mutation causing hATTR-CM is a valine to isoleucine substitution at amino acid 122 (V122I) that has been identified in 3.5% of African Americans and in 10% of African Americans older than 65 with severe congestive heart failure (Buxbaum JN and Ruberg FL, 2017). Increased incidence of the V122I mutation has been identified in a contiguous group of West African countries, the geographic origin of much of the slave trade (Jacobsen, DR et al, 2016). In African Americans, the allele frequency is 0.0173, compared to 0.0253 in Western Africans. By contrast, the allele frequency in patients who are not of African descent is vanishingly low (Yamashita T et al, 2009).
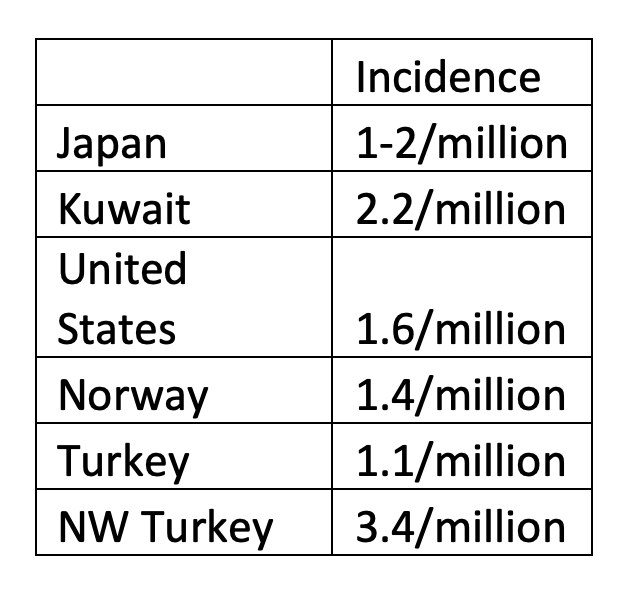
Like KD, there has been a great deal of research and awareness of TA in Japan, which may account for the reported data. There is increasing recognition of TA in Europe and elsewhere. Because of the rarity of the disease, exact data are difficult to interpret: the most frequently cited study of TA in the United States is from Olmstead County, Minnesota from 1985: incidence was found to be 2.6 x 106/year (Hall S et al, 1985). Since I’m mixing prevalence and incidence, here’s a table to give a bit of context (Onen F et al, 2017). Based on these data, it should be clear that TA should be considered in patients with vasculitis of the aorta and its major branches.


Previously, tissue biopsy was required for diagnosis (and remains the gold standard); however, nuclear scintigraphy with bone-avid tracers is a noninvasive method of diagnosis and should be considered in patients over 60 with congestive heart failure who are not significantly hypertensive (suggesting that their heart failure is not due to hypertension). It is likely that detection of hATTR-CM will increase with the use of this noninvasive diagnostic method.
Death from hATTR-CM usually occurs 2 to 6 years after diagnosis. A recently approved drug, tafamidis, binds to the thyroxine-binding sites and stabilizes the homotetramer which reduces dissociation and slows disease progression. Tafamidis areduces all-cause mortality (number needed to treat, 7.5 to prevent 1 death) as well as cardiac-associated hospitalizations (number needed to treat, 4 to prevent 1 hospitalization): at a cost of $225,000 per year (Gurwitz JH and Maurer MS, 2020; Maurer MS et al, 2018). Although hATTR-CM is rare in the general population, the increased incidence in the African American population which, disproportionately lacks access to care and private health insurance, increases the health burden in this population.
In the 11th edition, we removed the term “black”. The choice of replacement term is challenging. Since this is a mutation, we ideally would focus on ancestry, making a link to “individuals of African descent”. However, this allele is most common in West Africa and the increased incidence in the United States is because this part of Africa was the origin for enslaved people in our country’s history. We would not expect all “people of African descent” (i.e., anywhere in Africa) to have an increased risk of this allele; remember, there is more genetic diversity in Africa than anywhere else in the world. Ideally, we would have specified this population more clearly, but it was already a very contentious discussion…
Buxbaum JN, Ruberg FL. Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med. 2017 Jul;19(7):733-742. doi: 10.1038/gim.2016.200. Epub 2017 Jan 19. PMID: 28102864; PMCID: PMC5509498.
Damrauer SM, Chaudhary K, Cho JH, Liang LW, Argulian E, Chan L, Dobbyn A, Guerraty MA, Judy R, Kay J, Kember RL, Levin MG, Saha A, Van Vleck T, Verma SS, Weaver J, Abul-Husn NS, Baras A, Chirinos JA, Drachman B, Kenny EE, Loos RJF, Narula J, Overton J, Reid J, Ritchie M, Sirugo G, Nadkarni G, Rader DJ, Do R. Association of the V122I Hereditary Transthyretin Amyloidosis Genetic Variant With Heart Failure Among Individuals of African or Hispanic/Latino Ancestry. JAMA. 2019 Dec 10;322(22):2191-2202. doi: 10.1001/jama.2019.17935. PMID: 31821430; PMCID: PMC7081752.
Gorevic PD, Prelli FC, Wright J, Pras M, Frangione B. Systemic senile amyloidosis. Identification of a new prealbumin (transthyretin) variant in cardiac tissue: immunologic and biochemical similarity to one form of familial amyloidotic polyneuropathy. J Clin Invest. 1989 Mar;83(3):836-43. doi: 10.1172/JCI113966. PMID: 2646319; PMCID: PMC303756.
Gurwitz JH, Maurer MS. Tafamidis-A Pricey Therapy for a Not-So-Rare Condition. JAMA Cardiol. 2020 Mar 1;5(3):247-248. doi: 10.1001/jamacardio.2019.5233. PMID: 31913401.
Hamidi Asl K, Nakamura M, Yamashita T, Benson MD. Cardiac amyloidosis associated with the transthyretin Ile122 mutation in a Caucasian family. Amyloid. 2001 Dec;8(4):263-9. doi: 10.3109/13506120108993823. PMID: 11791619.
Jacobson DR, Alexander AA, Tagoe C, Garvey WT, Williams SM, Tishkoff S, Modiano D, Sirima SB, Kalidi I, Toure A, Buxbaum JN. The prevalence and distribution of the amyloidogenic transthyretin (TTR) V122I allele in Africa. Mol Genet Genomic Med. 2016 Jul 14;4(5):548-56. doi: 10.1002/mgg3.231. PMID: 27652282; PMCID: PMC5023940.
Jacobson DR, Gorevic PD, Buxbaum JN. A homozygous transthyretin variant associated with senile systemic amyloidosis: evidence for a late-onset disease of genetic etiology. Am J Hum Genet. 1990 Jul;47(1):127-36. PMID: 2349941; PMCID: PMC1683748.
Jacobson DR, Pastore RD, Yaghoubian R, Kane I, Gallo G, Buck FS, Buxbaum JN. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997 Feb 13;336(7):466-73. doi: 10.1056/NEJM199702133360703. PMID: 9017939.
Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, Kristen AV, Grogan M, Witteles R, Damy T, Drachman BM, Shah SJ, Hanna M, Judge DP, Barsdorf AI, Huber P, Patterson TA, Riley S, Schumacher J, Stewart M, Sultan MB, Rapezzi C; ATTR-ACT Study Investigators. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018 Sep 13;379(11):1007-1016. doi: 10.1056/NEJMoa1805689. Epub 2018 Aug 27. PMID: 30145929.
Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin Amyloid Cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872-2891. doi:10.1016/j.jacc.2019.04.003
Schumacher J, Stewart M, Sultan MB, Rapezzi C; ATTR-ACT Study Investigators. Tafamidis Treatment for Patients with Transthyretin Amyloid Cardiomyopathy. N Engl J Med. 2018 Sep 13;379(11):1007-1016. doi: 10.1056/NEJMoa1805689. Epub 2018 Aug 27. PMID: 30145929.
Yamashita T, Hamidi Asl K, Yazaki M, Benson MD. A prospective evaluation of the transthyretin Ile122 allele frequency in an African-American population. Amyloid. 2005 Jun;12(2):127-30. doi: 10.1080/13506120500107162. PMID: 16011990.
Takayasu aortitis
Although historically associated with Japanese ethnicity and certain HLA haplotypes, Takayasu aortitis has a global distribution. (BP10 p 385).
Athough historically associated with Japanese ethnicity and certain HLA alleles, Takayasu arteritis has a global distribution. (BP11, p 295).
Like Kawasaki disease, Takayasu aortitis (TA) was initially described in Japan and the disease has a high prevalence there (up to 40 per 106) (Toshihiko N, 1996). However, as with many of the diseases described in Robbins, a high prevalence in a particular population does not exclude a high prevalence in other populations. For example, in Turkey (on the opposite side of Asia), prevalence is 33 per 106) and in Norway it is 25.6 per 106).
Hall S, Barr W, Lie JT, Stanson AW, Kazmier FJ, Hunder GG. Takayasu arteritis. A study of 32 North American patients. Medicine (Baltimore). 1985 Mar;64(2):89-99. PMID: 2858047.
Onen F, Akkoc N. Epidemiology of Takayasu arteritis. Presse Med. 2017 Jul-Aug;46(7-8 Pt 2):e197-e203. doi: 10.1016/j.lpm.2017.05.034. Epub 2017 Jul 26. PMID: 28756072.
Seyahi E. Takayasu arteritis: an update. Curr Opin Rheumatol. 2017 Jan;29(1):51-56. doi: 10.1097/BOR.0000000000000343. PMID: 27748689.
Toshihiko N. Current status of large and small vessel vasculitis in Japan. Int J Cardiol. 1996 Aug;54 Suppl:S91-8. doi: 10.1016/s0167-5273(96)88777-8. PMID: 9119531.
Chapter Seven: Environmental Diseases
Vitamin D deficiency
However, blacks may have a lower level of vitamin D production in the skin because of melanin pigmentation (perhaps a small price to pay for protection against UV-induced cancers). BP p 329
First a quick review of vitamin D formation: When UVB strikes the epidermis, 7-dehydrocholesterol is converted into previtamin D3, which is next converted into vitamin D3 (cholecalciferol). In the liver, vitamin D3 is hydroxylated to form 25-hydroxy-vitamin-D3. In the kidney, it is finally converted into the biologically active form: 1α,25-dihydroxyvitamin-D3. Laboratory testing analyzes for 25-hydroxy-vitamin-D3, not the biologically active form.
Serum vitamin D levels have declined across all populations in the United States, perhaps due to increased messaging about the link between sun exposure and skin cancer. Several studies have reported a high incidence of vitamin D deficiency in African Americans, Mexican Americans and Asian Americans (Ginde AA et al, 2009; Mitchell DM et al, 2012). In the study by Ginde and colleagues, nearly all (97%) African Americans and the vast majority of Mexican Americans (90%) were vitamin D deficient. A meta-analysis of studies from Africa also reported significant vitamin D deficiency in African populations (Mogire RM et al, 2019).
Melanin competes with 7-dehydrocholesterol for UVB photons and it might be predicted that individuals with darkly pigmented skin would therefore be vitamin D deficient. Once we begin looking at vitamin D deficiency in such individuals, the topic becomes increasingly racialized with sweeping statements about differences in the bone density of Blacks and Whites and measurements of various bones that recalls the skull measurements (in particular, cranial capacity) that justified so-called “scientific” racism (Finkelstein JS et al, 2002; Looker AC, 2002). I believe the researchers’ intentions were good, but it still gave me reasons for concern.
In addition to the degree of skin pigmentation, there are factors that relate to serum vitamin D. Many foods (e.g., milk, cereals) are fortified with vitamin D. Multivitamin supplements are another source of vitamin D. Sunscreen use, increasing latitude and increased time spent indoors also decrease vitamin D production.
The point has been made that despite having lower levels of serum vitamin D, there does not appear to be an increased risk of fracture (a downstream effect) in dark-skinned individuals (Powe CE et al, 2013). Bone fragility and osteoporosis are also multifactorial and relate to body mass index, calcium intake, weight-bearing exercise and diet. Powe and colleagues found differences in allele frequencies in the coding region of the vitamin D-binding protein in patients who self-identified as White and Black. In Black patients, this genetic variant resulted in lower serum levels of vitamin D-binding protein which, therefore, result in levels of bioavailable 25-hydroxyvitamin D similar to that measured in White populations. Powe did not test the skin reflectivity of the individuals who self-identified as white or black. It is important to recognize that in the United States there is large variation in skin color amongst those who self-identify as Black, some of these individuals have skin tones as light as those who self-identify as White (Lona-Durazo F et al, 2019; Ho BK et al, 2015) Thus, the difference in levels in bioavailable 25-hydroxyvitamin D in this study resulted from greater numbers of darker skinned individuals in the self-identified black group. The Powe study was conducted on individuals livings in Baltimore MD, but a similar study design conducted on self-identified blacks in West Virginia or Washington state may have found a much smaller difference, due to the greater percentage of European-descent in self-identified Blacks from those states (Batai K et al, 2021).
Vitamin D is not only critical for bone health (e.g., rickets, osteomalacia, osteoporosis), but has been implicated in numerous other conditions including cardiovascular disease, autoimmune disease and susceptibility to infections. The latter has increased recent interest in the context of COVID-19. The value of vitamin D supplementation for non-skeletal disease is unclear, though supplementation is generally considered to be low risk.
Batai K et al. Genetic loci associated with skin pigmentation in African Americans and their effects on vitamin D deficiency. PLoS Genet. 2021 Feb 18;17(2):e1009319. doi: 10.1371/journal.pgen.1009319. PMID: 33600456; PMCID: PMC7891745
Finkelstein JS, Lee ML, Sowers M, Ettinger B, Neer RM, Kelsey JL, Cauley JA, Huang MH, Greendale GA. Ethnic variation in bone density in premenopausal and early perimenopausal women: effects of anthropometric and lifestyle factors. J Clin Endocrinol Metab. 2002 Jul;87(7):3057-67. doi: 10.1210/jcem.87.7.8654. PMID: 12107201.
Ginde AA, Liu MC, Camargo CA Jr. Demographic differences and trends of vitamin D insufficiency in the US population, 1988-2004. Arch Intern Med. 2009 Mar 23;169(6):626-32. doi: 10.1001/archinternmed.2008.604. PMID: 19307527; PMCID: PMC3447083.
Harris SS. Vitamin D and African Americans. J Nutr. 2006 Apr;136(4):1126-9. doi: 10.1093/jn/136.4.1126. PMID: 16549493.
Ho BK, Robinson JK. Color bar tool for skin type self-identification: A cross-sectional study. J Am Acad Dermatol. 2015 Aug;73(2):312-3.e1. doi: 10.1016/j.jaad.2015.05.024. PMID: 26183973; PMCID: PMC4506490.
The Lancet Diabetes Endocrinology. Vitamin D and COVID-19: why the controversy? Lancet Diabetes Endocrinol. 2021 Feb;9(2):53. doi: 10.1016/S2213-8587(21)00003-6. Epub 2021 Jan 11. PMID: 33444566.
Lona-Durazo F et al. Meta-analysis of GWA studies provides new insights on the genetic architecture of skin pigmentation in recently admixed populations. BMC Genet. 2019 Jul 17; 20(1):59. doi: 10.1186/s12863-019-0765-5. PMID: 31315583; PMCID: PMC6637524
Looker AC. The skeleton, race, and ethnicity. J Clin Endocrinol Metab. 2002 Jul;87(7):3047-50. doi: 10.1210/jcem.87.7.8779. PMID: 12107199.
Mitchell DM, Henao MP, Finkelstein JS, Burnett-Bowie SA. Prevalence and predictors of vitamin D deficiency in healthy adults. Endocr Pract. 2012 Nov-Dec;18(6):914-23. doi: 10.4158/EP12072.OR. PMID: 22982792; PMCID: PMC3755751.
Mogire RM, Mutua A, Kimita W, Kamau A, Bejon P, Pettifor JM, Adeyemo A, Williams TN, Atkinson SH. Prevalence of vitamin D deficiency in Africa: a systematic review and meta-analysis. Lancet Glob Health. 2020 Jan;8(1):e134-e142. doi: 10.1016/S2214-109X(19)30457-7. Epub 2019 Nov 27. PMID: 31786117; PMCID: PMC7024961.
Powe CE, Evans MK, Wenger J, Zonderman AB, Berg AH, Nalls M, Tamez H, Zhang D, Bhan I, Karumanchi SA, Powe NR, Thadhani R. Vitamin D-binding protein and vitamin D status of black Americans and white Americans. N Engl J Med. 2013 Nov 21;369(21):1991-2000. doi: 10.1056/NEJMoa1306357. PMID: 24256378; PMCID: PMC4030388.
Sudden Infant Death syndrome
The etiology of SIDS is unknown and it is likely multifactorial. It has been proposed that three factors contribute to SIDS: 1) an underlying vulnerability (e.g., genetic polymorphism, brainstem anomaly); a triggering event (e.g., gastrointestinal illness, airflow obstruction due to bedding); and 3) a vulnerable developmental state.
Many of the risk factors overlap. For example, lower socioeconomic status is associated with cigarette smoking, shorter intergestational intervals and late or no prenatal care (Garrett BE et al, 2019; Klebanoff MA, 1988). Shorter intergestational age is associated with younger mothers with less education. In the table above, listing African-American and American Indian ethnicity as a factor associated with sudden infant death syndrome is not likely to result from a higher frequency of genetic risk variants in these groups. This disparity is likely due to a differential in the socioeconomic factors (resulting from structural racism) associated with risk in these socially defined groups (UpToDate simply lists “non-white race” as a risk factor).
There are cultural differences that may be relevant, however, such as the value placed on bedsharing or on favored bedding types. Furthermore over the past decade there has been a serious critique of bedsharing as a risk factor in SIDS, countering with an evolutionary perspective supporting the benefits of mother-infant cosleeping (McKenna et al, 2007; Bartick et al, 2018). “Race” cannot be used as a short cut to determine which information is provided to which patients.
Garrett BE, Martell BN, Caraballo RS, King BA. Socioeconomic Differences in Cigarette Smoking Among Sociodemographic Groups. Prev Chronic Dis. 2019 Jun 13;16:E74. doi: 10.5888/pcd16.180553. PMID: 31198164; PMCID: PMC6583815.
Goldstein RD, Kinney HC. Race, Ethnicity, and SIDS. Pediatrics. 2017 Jun;139(6):e20170898. doi: 10.1542/peds.2017-0898. Epub 2017 May 15. PMID: 28562296.
Goldstein RD, Trachtenberg FL, Sens MA, Harty BJ, Kinney HC. Overall Postneonatal Mortality and Rates of SIDS. Pediatrics. 2016 Jan;137(1). doi: 10.1542/peds.2015-2298. Epub 2015 Dec 2. PMID: 26634772.
Klebanoff MA. Short interpregnancy interval and the risk of low birthweight. Am J Public Health. 1988 Jun;78(6):667-70. doi: 10.2105/ajph.78.6.667. PMID: 3369598; PMCID: PMC1350279.
McKenna JJ, Ball HL, Gettler LT. Mother-infant cosleeping, breastfeeding and sudden infant death syndrome: what biological anthropology has discovered about normal infant sleep and pediatric sleep medicine. Am J Phys Anthropol. 2007; Suppl 45:133-61. doi: 10.1002/ajpa.20736. PMID: 18046747
Bartick M, Tomori C, Ball HL. Babies in boxes and the missing links on safe sleep: Human evolution and cultural revolution. Matern Child Nutr. 2018 Apr;14(2):e12544. doi: 10.1111/mcn.12544. Epub 2017 Oct 18. PMID: 29047226; PMCID: PMC6866223.
Parks SE, Erck Lambert AB, Shapiro-Mendoza CK. Racial and Ethnic Trends in Sudden Unexpected Infant Deaths: United States, 1995-2013. Pediatrics. 2017 Jun;139(6):e20163844. doi: 10.1542/peds.2016-3844. Epub 2017 May 15. PMID: 28562272; PMCID: PMC5561464.
Singh GK, Yu SM. Infant mortality in the United States: trends, differentials, and projections, 1950 through 2010. Am J Public Health. 1995 Jul;85(7):957-64. doi: 10.2105/ajph.85.7.957. PMID: 7604920; PMCID: PMC1615523.
Chapter 6: Neoplasia
This chapter does a good job of associating geography with increased cancer risk, as opposed to suggesting there is a biological component to race. For example: “Hepatocellular carcinoma is the most lethal cancer in many parts of Africa. Most evidence suggests that these geographic differences have environmental origins. For example, Nisei (second-generation Japanese living in the United States) have mortality rates for certain forms of cancer that are intermediate between those in natives of Japan and in Americans who have lived in the United States for many generations. The two rates come closer with each passing generation” and “Aflatoxin B1 is of interest because it is a naturally occurring agent produced by some strains of Aspergillus, a mold that grows on improperly stored grains and nuts. A strong correlation has been found between the dietary level of this food contaminant and the incidence of hepatocellular carcinoma in parts of Africa and Southeast Asia.”
Nasopharyngeal carcinoma
Nasopharyngeal carcinoma is a rare neoplasm that merits comment because of (1) the strong epidemiologic links to EBV and (2) the high frequency of this cancer among the Chinese, which raises the possibility of viral oncogenesis on a background of genetic susceptibility (BP10, p 546).
Nasopharyngeal carcinoma (NPC) is characterized by a unique geographic distribution and strong association with Epstein-Barr virus (EBV) infection. EBV is a ubiquitous pathogen and latently infects about 90% of the world’s population. Men are 2-3 times more likely than women to be diagnosed with NPC and risk increases with age. Histologically, NPC is classified as 1) keratinizing squamous cell carcinoma (SCC); (2) nonkeratinizing SCC; and (3) basaloid SCC.
Though extremely rare globally, NPC is fairly common in southern China, particularly in Guangdong province (Jia W-H et al, 2010) and patients of Cantonese origin. Within China, regional variation in incidence is up to 50-fold. Chinese people who emigrate to low-risk areas and their children have a lower risk of NPC, which suggests that environmental factors play a role. In Southeast Asia, incidence of NPC is linked to the extent of social admixture and intermarriage with southern Chinese. Other populations that are affected include indigenous populations in Southeast Asia, the Arctic, the Middle East and Northern Africa (Chang ET and Adami HO, 2006).
Well-established risk factors include diets high in salted fish/preserved foods (with high levels of nitrosamines), elevated antibody titers against EBV, family history of NPC and certain HLA genotypes (Guo X et al, 2009; Chang ET and Adami HO, 2006). Discrepant results have been seen for smoking, cooking with wood, traditional medicines, inhalants and exposure to solvents. The importance of family history could be due to shared genetic or cultural factors.
I looked into epidemiologic data on NPC in the United States but the literature is incredibly disappointing. It is mostly based on SEER Surveillance, Epidemiology, and End Results (SEER) data which lumps all Asians together (not to mention the challenges in their race assignation protocol). NPC is extremely rare in the U.S. (0.5 – 2 cases per 100,000). Although it is unlikely, therefore, that you will encounter a patient with this disease, your index of suspicion should be increased for patients from areas in which this tumor is endemic and who present with unexplained headache, cranial nerve involvement, nasal obstruction or neck mass (due to metastatic disease) (Saba NF et al, 2015). Patients may be asymptomatic for a long duration since the symptoms are not specific and the site is occult.
For the 11th edition, we broadened the discussion of involved populations: “This tumor is endemic in southern China, parts of Africa, and the Inuit population of the Arctic.” The goal here as to reflect the distribution of the disease and to avoid labeling a certain ethnicity as being particularly prone to a certain disease. This practice does not serve you or your future patients!
Chang ET, Adami HO. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2006 Oct;15(10):1765-77. doi: 10.1158/1055-9965.EPI-06-0353. PMID: 17035381.
Guo X, Johnson RC, Deng H, Liao J, Guan L, Nelson GW, Tang M, Zheng Y, de The G, O'Brien SJ, Winkler CA, Zeng Y. Evaluation of nonviral risk factors for nasopharyngeal carcinoma in a high-risk population of Southern China. Int J Cancer. 2009 Jun 15;124(12):2942-7. doi: 10.1002/ijc.24293. PMID: 19296536; PMCID: PMC4406046.
Jia WH, Luo XY, Feng BJ, Ruan HL, Bei JX, Liu WS, Qin HD, Feng QS, Chen LZ, Yao SY, Zeng YX. Traditional Cantonese diet and nasopharyngeal carcinoma risk: a large-scale case-control study in Guangdong, China. BMC Cancer. 2010 Aug 20;10:446. doi: 10.1186/1471-2407-10-446. PMID: 20727127; PMCID: PMC2931495.
Saba NF, Salama JK, Beitler JJ, Busse PM, Cooper JS, Jones CU, Koyfman S, Quon H, Ridge JA, Siddiqui F, Worden F, Yao M, Yom SS; Expert Panel on Radiation Oncology-Head and Neck Cancer. ACR Appropriateness criteria® for nasopharyngeal carcinoma. Head Neck. 2016 Jul;38(7):979-86. doi: 10.1002/hed.24423. Epub 2016 Apr 30. PMID: 27131050.
Acetaldehyde dehydrogenase deficiency
Most notably, about 50% of Asians express a defective form of acetaldehyde dehydrogenase. (BP10, p. 311)
Terminology is shifting away from referring to “defects” and towards using the phrases “pathologic variant” or “genetic variant” or “polymorphism”. In this case, a polymorphism in the ALDH2 gene results in the substitution of a lysine for a glutamic acid at position 487. The resultant protein is not as effective at metabolizing ethanol and, as a consequence, acetaldehyde levels increase, giving rise to facial flushing and nausea. Individuals with the mutant allele are less likely to become alcohol dependent (Luo, H-R et al, 2009); however, when individuals with this mutation consume alcohol, their upper digestive tract mucosa and gastric mucosa are exposed to about 2-fold and 5- to 6-fold higher concentrations of acetaldehyde, respectively, than individuals who do not have this mutation (Lachenmeier DW and Salaspuro M, 2017) and risk of oral, pharyngeal, esophageal and gastric carcinomas is greatly increased.
Based on allele frequency and SNP data, the origin of the ALDH2 variant appears to be Yunnan, south coastal and east coastal China, possible among the Pai-Yuei tribe, which lived in this area 2000 – 3000 years ago (Luo, H-R et al, 2009). Another group has suggested that the allele arose in central China among the Han Chinese (Li H et al, 2009). The high frequency of this allele in Korea and Japan is thought to be related to migration from southern China about 2300 years ago.

For the 11th edition of Robbins, we used more accurate terminology: Acetaldehyde metabolism differs between populations due to genetic variation. One polymorphism that originated in China causes acetaldehyde accumulation in individuals of East Asian descent (e.g., China, Korea, Japan). After ingesting alcohol, individuals with this allele experience flushing, tachycardia, and hyperventilation.
Lachenmeier DW, Salaspuro M. ALDH2-deficiency as genetic epidemiologic and biochemical model for the carcinogenicity of acetaldehyde. Regul Toxicol Pharmacol. 2017 Jun;86:128-136. doi: 10.1016/j.yrtph.2017.02.024. Epub 2017 Mar 1. PMID: 28257851.
Li H, Borinskaya S, Yoshimura K, Kal'ina N, Marusin A, Stepanov VA, Qin Z, Khaliq S, Lee MY, Yang Y, Mohyuddin A, Gurwitz D, Mehdi SQ, Rogaev E, Jin L, Yankovsky NK, Kidd JR, Kidd KK. Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann Hum Genet. 2009 May;73(Pt 3):335-45. doi: 10.1111/j.1469-1809.2009.00517.x. PMID: 19456322; PMCID: PMC2846302.
Luo HR, Wu GS, Pakstis AJ, Tong L, Oota H, Kidd KK, Zhang YP. Origin and dispersal of atypical aldehyde dehydrogenase ALDH2487Lys. Gene. 2009 Apr 15;435(1-2):96-103. doi: 10.1016/j.gene.2008.12.021. Epub 2009 Jan 29. PMID: 19393179.
Shin MJ, Cho Y, Davey Smith G. Alcohol Consumption, Aldehyde Dehydrogenase 2 Gene Polymorphisms, and Cardiovascular Health in Korea. Yonsei Med J. 2017 Jul;58(4):689-696. doi: 10.3349/ymj.2017.58.4.689. PMID: 28540979; PMCID: PMC5447097.
Chapter 8: Blood Vessels
Hypertension
“The prevalence of pathologic effects of high blood pressure increases with age and is also higher in African Americans.” (BP10 p 366)
According to the CDC, high blood pressure has the following prevalence: non-Hispanic black adults (54%), non-Hispanic white adults (46%), non-Hispanic Asian adults (39%), and Hispanic adults (36%) (https://millionhearts.hhs.gov/data-reports/hypertension-prevalence.html).
Hypertension is undoubtedly multifactorial and due to a combination of genetic and environmental/social factors. African patients in sub-Saharan Africa do not have a similar incidence of hypertension as that seen in African Americans, which suggests that environmental/social factors may be more significant (Cooper, R.S., et al, 2005; Kearney, P.M., et al, 2005). Furthermore, studies in Africa have shown that when patients move from a traditional rural lifestyle to an urban, industrialized environment, blood pressure levels rise (Poulter NR et al, 1990). In addition, within one generation, Western African immigrants to the United States take on the disease patterns observed amongst African Americans (Read, Emerson, and Tarlov 2005).
In the media, the idea of a “slave hypertension gene” once caught popular attention, though there is no scientific support (for an excellent review, read Lujan, H.K and DiCarlo, S.E., Adv. Physiol. Educ. 2018). The initial iteration of this hypothesis was that, due to arid and salt-poor conditions in Africa, there was selection pressure for salt retention – which would account for present-day salt retention in African Americans. The science of this hypothesis is weak since a) African populations did not have limited access to salt (Graves 2005a) and b) Africans actually have less hypertension than African Americans. The theory then developed to suggest that the horrific conditions aboard slave ships (e.g., dehydration, diarrhea, vomiting) selected for survivors who were better at retaining sodium. The hard work and privations of slavery additionally selected for sodium retention. However, this theory is not supported by basic facts of natural selection: the mortality rate of the middle passage would need to be much higher than historical data suggest to exert this degree of selection pressure in such a short time (Graves JL, 2005b). Furthermore, such a population bottleneck would be associated with an increased prevalence of recessive diseases in this population, which is not seen. Similarly, it is far more likely that selection would have acted to increase salt retention is North Western Europeans who migrated from a temperate to a subtropical environment in the Americas (e.g. Virginia, Carolinas, Georgia), then it would have acted on a tropical adapted Africans moving to those same regions.
What does account for the increased prevalence of hypertension in African Americans? “Lifestyle” factors such as obesity, smoking, alcohol use, physical exercise and salt intake have been suggested as possible explanations, but these do not appear to entirely account for the observed disparity. The “neighborhood” effect (i.e., examining populations from inner city neighborhoods or areas with extreme poverty) has been correlated with increased hypertension (Diez-Roux, A.V. et al, 2007; Harburg, E. et al, 1973) which also may relate to increased toxin burden (e.g., lead exposure). The effects of systemic racism are difficult to quantify since the consequences are protean (for example, the experience of discrimination may lead to unhealthy behaviors); furthermore, the data are challenging to interpret (Forde, A.T. et al. 2020; Hicken, M.T. et al., 2014; Krieger N et al, 1996).
For a detailed discussion of hypertension in African Americans, I highly recommend pages 124-136 in Dr. Joseph Graves’ book, “The Race Myth: Why We Pretend Race Exists in America.”
Some of the environmental and socioeconomic factors that affect African Americans also have an impact on Hispanics, American Indians/Alaskan Natives and Asian Americans. The Hispanic population in the United States is incredibly diverse and includes individuals from more than 20 countries and representing a variety of cultural, social and economic characteristics that have an impact on health (Campos CL et al, 2019; Elfassy T et al, 2020). Aggregation of data across these different groups has complicated data analysis and, in general, the impact of hypertension in Hispanic Americans is not well documented and additional research is necessary to further refine our understanding. American Indians/Alaskan Natives are about 1-% more likely to be hypertensive than individuals of European descent which may be linked to higher rates of cigarette smoking and obesity (https://minorityhealth.hhs.gov/omh/browse. aspx?lvl=4&lvlid=34).
Geography, too, plays a role in hypertension prevalence. In fact, the difference between California and Oklahoma is greater than the difference between the CDC data of high blood pressure between non-Hispanic black adults and non-Hispanic white adults (˜14% vs 8%):

The real issue here is resources for care: in the United States, EGFR testing is widely available. The College of American Pathologists recommends that “EGFR molecular testing should be used to select patients for EGFR-targeted TKI therapy, and patients with lung adenocarcinoma should not be excluded from testing on the basis of clinical characteristics [e.g., “race”, ethnicity]” (Lindemann NI et al, 2013). In resource-rich countries, so-called “race” and ethnicity should not be the factor that determines if mutational testing is performed. However, in lower income countries where testing may not be universally available, cost-benefit analysis may result in some patients not being tested. Resources also come into play when it comes to actually paying for tyrosine kinase inhibitors which can cost up to $10,000/month in the United States (Olszewski AJ et al, 2018).
Figure 3 | The global distribution of the β-thalassaemia mutations. The common mild mutations are shown in bold. β-thalassaemia also occurs in the regions shaded in grey, but little is known about its molecular pathology in these areas.

The mechanism by which the thalassemias protect against malaria is not clearly defined. Two mechanisms that have been suggested include enhanced antibody binding with increased clearance of infected red cells and increased phagocytosis of infected red cells (Kariuki SN et al, 2020; Ayi K et al, 2004; Yuthavong Y et al, 1990; Yuthavong Y et al, 1988; Luzzi GA et al, 1991).
The wording remains essentially the same in Robbins & Kumar Basic Pathology, 11th edition since the original material was correct.
Ayi K, Turrini F, Piga A, Arese P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: a common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood. 2004 Nov 15;104(10):3364-71. doi: 10.1182/blood-2003-11-3820. Epub 2004 Jul 27. PMID: 15280204
Harteveld CL, Higgs DR. Alpha-thalassaemia. Orphanet J Rare Dis. 2010 May 28;5:13. doi: 10.1186/1750-1172-5-13. PMID: 20507641; PMCID: PMC2887799.
Kariuki SN, Williams TN. Human genetics and malaria resistance. Hum Genet. 2020 Jun;139(6-7):801-811. doi: 10.1007/s00439-020-02142-6. Epub 2020 Mar 4. PMID: 32130487; PMCID: PMC7271956.
Luzzi GA, Merry AH, Newbold CI, Marsh K, Pasvol G, Weatherall DJ. Surface antigen expression on Plasmodium falciparum-infected erythrocytes is modified in alpha- and beta-thalassemia. J Exp Med. 1991 Apr 1;173(4):785-91. doi: 10.1084/jem.173.4.785. PMID: 2007853; PMCID: PMC2190806.
Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001 Apr;2(4):245-55. doi: 10.1038/35066048. PMID: 11283697.
Yuthavong Y, Bunyaratvej A, Kamchonwongpaisan S. Increased susceptibility of malaria-infected variant erythrocytes to the mononuclear phagocyte system. Blood Cells. 1990;16(2-3):591-7. PMID: 2098019.
Yuthavong Y, Butthep P, Bunyaratvej A, Fucharoen S, Khusmith S. Impaired parasite growth and increased susceptibility to phagocytosis of Plasmodium falciparum infected alpha-thalassemia or hemoglobin Constant Spring red blood cells. Am J Clin Pathol. 1988 Apr;89(4):521-5. doi: 10.1093/ajcp/89.4.521. PMID: 3281435.
Glucose-6-phosphate dehydrogenase (G6PD) deficiency
One is G6PD A−, which is carried by approximately 10% of black males in the United States. (BP10, p 450).
G6PD deficiency is primarily found in areas where malaria is/was endemic. More than 200 (BP says >400) genetic variants of this disease that have been categorized into 5 classes by the WHO (class I worst, class IV, V silent) (UpToDate). There are named variants that are more prevalent in certain populations such as G6PD Mediterranean (563C>T) (class II) is more common in Mediterranean and Middle Eastern populations; G6PD A- (class III) which is most common in people with African ancestry; and G6PD Canton (class II or III) which is most prevalent in China.
The mechanism by which G6PD deficiency protects against malaria is not known. The link between malaria and the disease is primarily epidemiological, but also observational: lower parasite infection burden has been seen in hemizygous males and heterozygous females than wild type patients (Bienzle U et al, 1972; Ruwende C et al, 1995; Manzurano A et al, 2015). Malaria parasites break down hemoglobin shortly after entering the cell which can lead to an increased concentration of oxidized iron which may be toxic to the parasite. Since G6PD is part of the pathway for generating reduced glutathione (an antioxidant), it is possible that deficient states contribute to parasite killing (Tripathy V and Reddy BM, 2007).
Hemolytic anemia in the setting of G6PD deficiency is due to oxidative stress and can occur secondary to dietary indiscretion (e.g., fava beans), infection, and certain drugs. There is no absolute agreement on which drugs are safe and which ones are absolutely contraindicated since the latter, in particular, depends on the degree of hemolysis and the clinical setting. Of particular note are the antimalarial drugs chloroquine and hydroxychloroquine which have been considered in the treatment of COVID-19. Other antimalarial drugs such as primaquine can also cause hemolytic anemia which can manifest in homozygous and heterozygous women, depending on the specific mutation.
In the 11th edition, we removed the sentence referring to the 10% incidence in “black males”. This wording is problematic since a) it uses “color” language which we have removed from this edition and b) the reference should be to ancestry since this is a genetic disease. Furthermore, we wanted to emphasize that this is a disease found in numerous populations, not just those of African descent. The new edition says: In the case of the G6PD A- variant, which is common in areas of Africa where malaria is endemic, the half-life of the variant is only modestly decreased. As a result, only older red cells are susceptible to lysis. Because the marrow compensates for the anemia by increasing its production of new red cells with adequate levels of G6PD, the hemolysis abates even if the drug exposure continues. Other variants such as G6PD Mediterranean, found mainly in the Middle East, produce more marked enzyme deficiency and as a result the hemolysis that occurs on exposure to oxidants is more severe.
Bienzle U, Ayeni O, Lucas AO, Luzzatto L. Glucose-6-phosphate dehydrogenase and malaria. Greater resistance of females heterozygous for enzyme deficiency and of males with non-deficient variant. Lancet. 1972 Jan 15;1(7742):107-10. doi: 10.1016/s0140-6736(72)90676-9. PMID: 4108978.
Manjurano A, Sepulveda N, Nadjm B, Mtove G, Wangai H, Maxwell C, Olomi R, Reyburn H, Riley EM, Drakeley CJ, Clark TG; MalariaGEN Consortium. African glucose-6-phosphate dehydrogenase alleles associated with protection from severe malaria in heterozygous females in Tanzania. PLoS Genet. 2015 Feb 11;11(2):e1004960. doi: 10.1371/journal.pgen.1004960. PMID: 25671784; PMCID: PMC4335500.
Ruwende C, Khoo SC, Snow RW, Yates SN, Kwiatkowski D, Gupta S, Warn P, Allsopp CE, Gilbert SC, Peschu N, et al. Natural selection of hemi- and heterozygotes for G6PD deficiency in Africa by resistance to severe malaria. Nature. 1995 Jul 20;376(6537):246-9. doi: 10.1038/376246a0. PMID: 7617034
Tripathy V, Reddy BM. Present status of understanding on the G6PD deficiency and natural selection. J Postgrad Med. 2007 Jul-Sep;53(3):193-202. doi: 10.4103/0022-3859.33867. PMID: 17699998.
Multiple myeloma
The median age at diagnosis is 70 years, and it [multiple myeloma] is more common in males and in people of African origin. BP10 p 472
In the United States, multiple myeloma (MM) is the most common hematologic malignancy in African Americans and occurs at about twice the rate as in Americans of European descent (Ries LAG et al, 2002). African Americans tend to present at a younger age, with increased numbers of comorbidities, with a lower hemoglobin and with increased serum lactate dehydrogenase as compared to patients of European descent (Waxman AJ et al, 2010, Ailawadhi S et al, 2018). These laboratory values are consistent with greater disease burden.
Disease incidence in Africa is much lower; however, this is likely due to underdiagnosis as opposed to genetic or environmental factors. Two studies in the Caribbean of patients with MM who were of African descent demonstrated a prevalence of 3.1-5/100,000 which is comparable to the reported rate in individuals of European descent (4/100,000) during the same time period (Besson C et al, 2001; Nossent JC et al, 1993; Ries LAG et al, 2002) though Shirley and colleagues found much higher rates in the United Kingdom (8.3 – 8.6/100,000 in patients of African descent compared to 3.7/100,000 in patients of European descent) as did Waxman and colleagues (11.0 and 4.9/100,000 for patients of African and European descent, respectively) (Waxman AJ et al, 2010; Shirley MH et al, 2013). The reason for these disparities in the incidence data is not clear.
It is also not clear why there is an observed disparity in the incidence of MM between African Americans and Americans of European descent. There is likely a genetic component to MM based on familial clustering, increased risk in first-degree relatives and association with certain HLA haplotypes (Benjamin M et al, 2003). While a number of environmental factors (e.g., exposure to toxins and radiation, diet, tobacco use, alcohol use) have been evaluated epidemiologically, due to the rarity of MM, most studies lack sufficient statistical power for meaningful conclusions (Alexander DD et al, 2007). Obesity appears to be a consistent risk factor for MM (Hoffmann JN, 2013).
Most hematologic malignancies require sophisticated tests for diagnosis such as flow cytometry and molecular analysis; multiple myeloma (MM) is unusual in that the diagnosis can be made using relatively common and inexpensive modalities (i.e., basic metabolic panel, complete blood count with differential, serum calcium, serum and urine electrophoresis and radiologic evaluation of the skeleton) (Cowan AJ et al, 2018). Nonetheless, underdiagnosis is believed to account for the low reported incidence in lower and middle-income countries. In these regions,
there is often a delay in diagnosis and lack of access to appropriate and affordable care, which accounts for the worse prognosis in this setting, despite advances in outcome in economically privileged countries (Omoti CE et al, 2007; Nwabuko OC et al, 2017).
Lack of access to appropriate care is also seen in the United States. Prognosis has improved for patients with MM due to increased use of novel therapies (e.g., lenalidomide, bortezomib) and autologous stem cell transplant. However, it has been shown that patients of African and Hispanic descent are less likely to receive these therapies (Ailawadhi S et al, 2017; Abouzaid S et al, 2016). Interestingly, a recent study has shown in the VA system that when patients with MM were given equal access to treatments (e.g., novel therapeutics, stem cell transplant), African Americans had improved survival compared to patients of European descent (Fillmore NR et al, 2019).
It is difficult to discuss this material accurately. Since this is not a single-gene disease and, instead, is due to multiple loci and environmental factors such as exposure to toxins, dietary issues, etc., it would be best to use socially defined race when comparing these disparities, as opposed to ancestry: therefore, MM is higher in African Americans than in European Americans as opposed to being higher in people of African descent compared to European descent. But when we really look at the data, the clinical significance of emphasizing this difference is questionable: we’re talking here about 12 in 100,000 vs 6 in 100,000. With an incidence this low, positive predictive value for laboratory tests is essentially zero. This is not how we diagnose disease: we don’t gather 100,000 African Americans and 100,000 European Americans and say, “12 of you have this and 6 of you have this and we’re going to figure out who it is”! We look at each patient, evaluate their symptoms, radiology and laboratory tests. Nonetheless, I was unable to get this factoid removed from the 11th edition.
Abouzaid, S., K. Parikh, Z. Y. Zhou, Z. Zhou, W. Tang, J. Xie, et al. 2016. Disparities in treatment patterns and outcomes between Caucasian and African American patients with multiple myeloma (MM). J. Clin. Oncol. 34:802
Ailawadhi S, Frank RD, Advani P, Swaika A, Temkit M, Menghani R, Sharma M, Meghji Z, Paulus S, Khera N, Hashmi SK, Paulus A, Kakar TS, Hodge DO, Colibaseanu DT, Vizzini MR, Roy V, Colon-Otero G, Chanan-Khan AA. Racial disparity in utilization of therapeutic modalities among multiple myeloma patients: a SEER-medicare analysis. Cancer Med. 2017 Dec;6(12):2876-2885. doi: 10.1002/cam4.1246. Epub 2017 Nov 3. PMID: 29105343; PMCID: PMC5727310.
Ailawadhi S, Jacobus S, Sexton R, Stewart AK, Dispenzieri A, Hussein MA, Zonder JA, Crowley J, Hoering A, Barlogie B, Orlowski RZ, Rajkumar SV. Disease and outcome disparities in multiple myeloma: exploring the role of race/ethnicity in the Cooperative Group clinical trials. Blood Cancer J. 2018 Jul 6;8(7):67. doi: 10.1038/s41408-018-0102-7. PMID: 29980678; PMCID: PMC6035273.
Alexander DD, Mink PJ, Adami HO, Cole P, Mandel JS, Oken MM, Trichopoulos D. Multiple myeloma: a review of the epidemiologic literature. Int J Cancer. 2007;120 Suppl 12:40-61. doi: 10.1002/ijc.22718. PMID: 17405120
Benjamin M, Reddy S, Brawley OW. Myeloma and race: a review of the literature. Cancer Metastasis Rev. 2003 Mar;22(1):87-93. doi: 10.1023/a:1022268103136. PMID: 12716040.
Besson C, Gonin C, Brebion A, Delaunay C, Panelatti G, Plumelle Y. Incidence of hematological malignancies in Martinique, French West Indies, overrepresentation of multiple myeloma and adult T cell leukemia/lymphoma. Leukemia. 2001 May;15(5):828-31. doi: 10.1038/sj.leu.2402040. PMID: 11368445.
Fiala MA, Wildes TM. Racial disparities in treatment use for multiple myeloma. Cancer. 2017 May 1;123(9):1590-1596. doi: 10.1002/cncr.30526. Epub 2017 Jan 13. PMID: 28085188; PMCID: PMC5400674.
Fillmore NR, Yellapragada SV, Ifeorah C, Mehta A, Cirstea D, White PS, Rivero G, Zimolzak A, Pyarajan S, Do N, Brophy M, Munshi NC. With equal access, African American patients have superior survival compared to white patients with multiple myeloma: a VA study. Blood. 2019 Jun 13;133(24):2615-2618. doi: 10.1182/blood.2019000406. Epub 2019 Apr 19. PMID: 31003998; PMCID: PMC6566591.
Hofmann JN, Moore SC, Lim U, Park Y, Baris D, Hollenbeck AR, Matthews CE, Gibson TM, Hartge P, Purdue MP. Body mass index and physical activity at different ages and risk of multiple myeloma in the NIH-AARP diet and health study. Am J Epidemiol. 2013 Apr 15;177(8):776-86. doi: 10.1093/aje/kws295. Epub 2013 Mar 29. PMID: 23543160; PMCID: PMC3668425.
Kaya H, Peressini B, Jawed I, Martincic D, Elaimy AL, Lamoreaux WT, Fairbanks RK, Weeks KA, Lee CM. Impact of age, race and decade of treatment on overall survival in a critical population analysis of 40,000 multiple myeloma patients. Int J Hematol. 2012 Jan;95(1):64-70. doi: 10.1007/s12185-011-0971-z. Epub 2011 Dec 9. PMID: 22160833.
Kazandjian D, Hill E, Hultcrantz M, Rustad EH, Yellapantula V, Akhlaghi T, Korde N, Mailankody S, Dew A, Papaemmanuil E, Maric I, Kwok M, Landgren O. Molecular underpinnings of clinical disparity patterns in African American vs. Caucasian American multiple myeloma patients. Blood Cancer J. 2019 Feb 4;9(2):15. doi: 10.1038/s41408-019-0177-9. PMID: 30718460; PMCID: PMC6361959.
Nossent JC, Winkel CN, van Leeuwen JC. Multiple myeloma in the Afro-Caribbean population of Curaçao. Neth J Med. 1993 Dec;43(5-6):210-4. PMID: 8107926.
Nwabuko OC, Igbigbi EE, Chukwuonye II, Nnoli MA. Multiple myeloma in Niger Delta, Nigeria: complications and the outcome of palliative interventions. Cancer Manag Res. 2017 May 22;9:189-196. doi: 10.2147/CMAR.S126136. PMID: 28579833; PMCID: PMC5446965.
Omoti CE, Omuemu CE. Multiple myeloma: a ten year study of survival and therapy in a developing nation. J Pak Med Assoc. 2007 Jul;57(7):341-4. PMID: 17867255.
Perrotta C, Staines A, Codd M, Kleefeld S, Crowley D, T' Mannetje A, Becker N, Brennan P, De Sanjosé S, Foretova L,
Maynadié M, Nieters A, Boffetta P, Cocco P. Multiple Myeloma and lifetime occupation: results from the EPILYMPH study. J Occup Med Toxicol. 2012 Dec 14;7(1):25. doi: 10.1186/1745-6673-7-25. PMID: 23241100; PMCID: PMC3557218.
Pierre A, Williams TH. African American Patients With Multiple Myeloma: Optimizing Care to Decrease Racial Disparities. Clin J Oncol Nurs. 2020 Aug 1;24(4):439-443. doi: 10.1188/20.CJON.439-443. PMID: 32678364.
Pulte D, Redaniel MT, Brenner H, Jeffreys M. Changes in survival by ethnicity of patients with cancer between 1992-1996 and 2002-2006: is the discrepancy decreasing? Ann Oncol. 2012 Sep;23(9):2428-2434. doi: 10.1093/annonc/mds023. Epub 2012 Mar 6. PMID: 22396445.
Pulte D, Redaniel MT, Brenner H, Jansen L, Jeffreys M. Recent improvement in survival of patients with multiple myeloma: variation by ethnicity. Leuk Lymphoma. 2014 May;55(5):1083-9. doi: 10.3109/10428194.2013.827188. Epub 2013 Sep 3. PMID: 23879201.
Ries LAG, Eisner MP, Kosary CL, Hankey BF, Miller BA, Clegg L, and Edwards BK: SEER Cancer Statistics Review, 1973–1999. http://seer.cancer.gov/csr/1973_1999/, 2002. Bethesda, MD, National Cancer Institute, 1999.
Shirley MH, Sayeed S, Barnes I, Finlayson A, Ali R. Incidence of haematological malignancies by ethnic group in England, 2001-7. Br J Haematol. 2013 Nov;163(4):465-77. doi: 10.1111/bjh.12562. Epub 2013 Sep 14. PMID: 24033296.
Smith CJ, Ambs S, Landgren O. Biological determinants of health disparities in multiple myeloma. Blood Cancer J. 2018 Aug 28;8(9):85. doi: 10.1038/s41408-018-0118-z. PMID: 30190459; PMCID: PMC6127236.
Follicular lymphoma
This relatively common tumor [follicular lymphoma] constitutes 40% of the adult NHLs in the United States. Like CLL/SLL, it occurs much less frequently in Asian populations.
One of the challenges in assessing the literature regarding disease incidence in “Asians” and “Asian Americans” is that these groups are extremely heterogeneous and span multiple ethnicities and a broad geographical area. I have tried to identify studies that were more specific in their analysis (e.g., Carreon JD et al, 2008; Yang C et al, 1991). The reduced incidence of follicular lymphoma (FL) and chronic lymphocytic leukemia /small lymphocytic lymphoma (CLL/SLL) in patients of Asian descent is well documented (Morton LM et al, 2006; Clark CA et al, 2011; Yang SM et al, 2015; Carreon JD et al, 2008)). CLL/SLL is about 5 to 10 times less common in Asian populations (Yang SM et al, 2015) with a rate of 0.5/100,000 in one study of the Han Chinese (Yang C et al, 1991).
SLL/CLL in Asians tends to occur in younger patients and have more atypical morphologic and immunologic features (e.g., prolymphocytic leukemia variant, increased expression of CD22, CD20 and FCM7) than patients of primarily European descent (Yang SM et al, 2015; Tomomatsu J, 2010; Jang MA et al, 2013). While prognostic factors of survival are similar, CLL appears to be more aggressive in patients of Asian descent than in patients of European descent (Yang SM et al, 2015).
Both environmental and genetic factors have been considered, though the latter, as yet undefined, are considered more important. A number of studies have examined HLA subtype and risk; however, the majority have been in European populations (Zhong C et al, 2019).
There has been considerable research into the contribution of environmental effects (e.g., birthplace, household crowding, childhood infections) in the etiology of FL and CLL/SLL (Bracci PM et al, 2006; Clark CA et al, 2001; Herrinton LJ et al, 1996; Parodi S et al, 2019),. In one study, where the country of birth was not specified, rates of follicular lymphoma and CLL/SLL in foreign-born Asian Americans are lower than in native-born Asian Americans (Clarke CA et al, 2011). In another study, where country of birth was specified, there was an increased incidence in follicular lymphoma in Chinese- and Japanese Americans born in the United States compared to foreign-born Chinese- and Japanese Americans; by contrast, in CLL/SLL, the increased incidence in US-born individuals was seen in Chinese Americans, but not Japanese- or Filipino Americans (Herrinton LJ t al, 1996).
In the 11th edition of Robbins, it says, “This relatively common tumor constitutes approximately 30% of NHLs in adults in the United States. Like CLL/SLL, it occurs less frequently in Asian populations.” I was unable to sway the editor of this chapter, though the data presented above present the issue fairly clearly: we cannot talk about differences in specific populations if the populations themselves are not specific.
Bracci PM, Dalvi TB, Holly EA. Residential history, family characteristics and non-Hodgkin lymphoma, a population-based case-control study in the San Francisco Bay Area. Cancer Epidemiol Biomarkers Prev. 2006 Jul;15(7):1287-94. doi: 10.1158/1055-9965.EPI-06-0066. PMID: 16835325.
Carreon JD, Morton LM, Devesa SS, et al. Incidence of lymphoid neoplasms by subtype among six Asian ethnic groups in the United States, 1996-2004. Cancer Causes Control. 2008;19(10):1171-1181. doi:10.1007/s10552-008-9184-z
Clarke CA, Glaser SL, Gomez SL, Wang SS, Keegan TH, Yang J, Chang ET. Lymphoid malignancies in U.S. Asians: incidence rate differences by birthplace and acculturation. Cancer Epidemiol Biomarkers Prev. 2011 Jun;20(6):1064-77. doi: 10.1158/1055-9965.EPI-11-0038. Epub 2011 Apr 14. PMID: 21493873; PMCID: PMC3111874.
Grulich AE, Vajdic CM. The epidemiology of non-Hodgkin lymphoma. Pathology. 2005 Dec;37(6):409-19. doi: 10.1080/00313020500370192. PMID: 16373224.
Herrinton LJ, Goldoft M, Schwartz SM, Weiss NS. The incidence of non-Hodgkin's lymphoma and its histologic subtypes in Asian migrants to the United States and their descendants. Cancer Causes Control. 1996 Mar;7(2):224-30. doi: 10.1007/BF00051298. PMID: 8740735.
Jang MA, Yoo EH, Kim K, Kim WS, Jung CW, Kim SH, Kim HJ. Chronic lymphocytic leukemia in Korean patients: frequent atypical immunophenotype and relatively aggressive clinical behavior. Int J Hematol. 2013 Mar;97(3):403-8. doi: 10.1007/s12185-013-1286-z. Epub 2013 Feb 12. PMID: 23400412.
Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006 Jan 1;107(1):265-76. doi: 10.1182/blood-2005-06-2508. Epub 2005 Sep 8. PMID: 16150940; PMCID: PMC1895348.
Parodi S, Seniori Costantini A, Crosignani P, Fontana A, Miligi L, Nanni O, Piro S, Ramazzotti V, Rodella S, Tumino R, Vindigni C, Vineis P, Stagnaro E. Childhood infectious diseases and risk of non-Hodgkin's lymphoma according to the WHO classification: A reanalysis of the Italian multicenter case-control study. Int J Cancer. 2020 Feb 15;146(4):977-986. doi: 10.1002/ijc.32393. Epub 2019 May 17. PMID: 31077355.
Shirley MH, Sayeed S, Barnes I, Finlayson A, Ali R. Incidence of haematological malignancies by ethnic group in England, 2001-7. Br J Haematol. 2013 Nov;163(4):465-77. doi: 10.1111/bjh.12562. Epub 2013 Sep 14. PMID: 24033296.
Tomomatsu J, Isobe Y, Oshimi K, Tabe Y, Ishii K, Noguchi M, Hirano T, Komatsu N, Sugimoto K. Chronic lymphocytic leukemia in a Japanese population: varied immunophenotypic profile, distinctive usage of frequently mutated IGH gene, and indolent clinical behavior. Leuk Lymphoma. 2010 Dec;51(12):2230-9. doi: 10.3109/10428194.2010.527403. Epub 2010 Nov 11. PMID: 21067444.
Wang SS, Abdou AM, Morton LM, Thomas R, Cerhan JR, Gao X, Cozen W, Rothman N, Davis S, Severson RK, Bernstein L, Hartge P, Carrington M. Human leukocyte antigen class I and II alleles in non-Hodgkin lymphoma etiology. Blood. 2010 Jun 10;115(23):4820-3. doi: 10.1182/blood-2010-01-266775. Epub 2010 Apr 12. PMID: 20385791; PMCID: PMC2890176.
Wu SJ, Chen YC, Lo WC, Chiang CJ, Lin CT, Chuang SS, Lai MS. Distinctive incidence patterns of follicular lymphoma in Taiwan: Implications of ethnic differences. Cancer Med. 2019 Apr;8(4):1899-1907. doi: 10.1002/cam4.2028. Epub 2019 Feb 21. PMID: 30791222; PMCID: PMC6488204.
Yang C, Zhang X. Incidence survey of leukemia in China. Chin Med Sci J. 1991 Jun;6(2):65-70. PMID: 1804379.
Yang SM, Li JY, Gale RP, Huang XJ. The mystery of chronic lymphocytic leukemia (CLL): Why is it absent in Asians and what does this tell us about etiology, pathogenesis and biology? Blood Rev. 2015 May;29(3):205-13. doi: 10.1016/j.blre.2014.12.001. Epub 2014 Dec 13. PMID: 25541495.
Zhong C, Cozen W, Bolanos R, Song J, Wang SS. The role of HLA variation in lymphoma aetiology and survival. J Intern Med. 2019 Aug;286(2):154-180. doi: 10.1111/joim.12911. Epub 2019 Jun 3. PMID: 31155783.
Chapter 12: Renal Pathology
Nephrosclerosis
This renal lesion [nephrosclerosis] alone rarely causes damage severe enough to produce renal failure, except in individuals from some, possibly genetically susceptible, groups, such as African Americans, in whom it may lead to uremia and death. BP10 p 570
Nephrosclerosis is defined as sclerosis of the small renal arteries, typically in response to hypertension. The arterioles show hyaline arteriolosclerosis, while fibrous intimal and medial thickening are seen in arteries, both of which lead to luminal narrowing. Downstream effects include tissue ischemia with consequent interstitial fibrosis, tubular atrophy and focal global glomerulosclerosis.
African Americans develop end-stage kidney disease (ESKD) about 3.5 times more frequently than European Americans: in 2010, about 0.1% of the African American population was diagnosed with ESKD, compared to 0.03% of European Americans (United States Renal Data System). Part of this risk is attributable to inheritance of two variants of the APOL1 allele, called G1 and G2 that evolved in Africa approximately 10,000 years ago (Genovese G et al, 2010). More than 50% of African Americans carry one of these two alleles, while 13% have a high-risk combination (either homozygous for one allele or compound heterozygous for both alleles: G1/G1, G1/G2 or G2/G2) (Drummer PD et al, 2015). This high-risk combination is associated with increased incidence of ESKD, focal segmental glomerulosclerosis and HIV-associated nephropathy. The high prevalence and short timeline since evolution of these alleles suggests a strong positive selective pressure…
Just as we have seen in sickle cell disease, in which a heterozygote state for HbS is protective against an infectious disease (i.e., malaria), these APOL1 variants are protective against sleeping sickness (African trypanosomiasis). African trypanosomiasis is caused by two variants of the parasite Trypanosoma brucei, one more prevalent in East Africa (Trypanosoma brucei rhodesiense) and one that is more prevalent in West Africa (T. b. gambiense) And, similar to what we saw in sickle cell disease, there is an increased incidence of these alleles in areas where the disease is prevalent (Cooper A et al, 2017). The G1 allele is associated with the asymptomatic carrier state when individuals are infected with T. b. gambiense (as opposed to clinically significant disease) but does not protect against T. b. rhodesiense. By contrast, individuals with the G2 allele were protected against infection with T.b. rhodesiense infection, but had increased risk of a more severe disease outcome when infected with T.b. gambiense. These associations are reflected in the geographic distribution of the two alleles (see right, Cooper A et al, 2017).
Risk of STDs correlates with 1) age at first sexual intercourse; 2) number of partners; 3) drug/alcohol use; 4) inconsistent condom use; 5) concurrent sexual relationships; 6) risky partners (e.g., partners who inject drugs); and 7) community infection rate.
In the United States, lower age at first sexual intercourse is a risk factor for increased numbers of sexual partners, increased likelihood of having risky sexual partners and increased likelihood for sexual intercourse under the influence of drugs or alcohol (Sandfort TG et al, 2008). Age at first sexual intercourse is lower for African Americans than European American and Hispanic males which increases the “window” of sexual exposure and also likely accounts for the higher number of reported sexual partners in this population (Hamilton DT and Morris M, 2015; Dariotis, J.K. et al, 2011).
A further cause of health disparity relates to actual control of high blood pressure. According to the CDC, of patients for whom blood pressure medication is recommended, blood pressure control as follows: non-Hispanic white adults (32%), non-Hispanic black adults (25%), non-Hispanic Asian adults (19%), and Hispanic adults (25%). (https://millionhearts.hhs.gov/data-reports/hypertension-prevalence.html).
In the 11th edition of Robbins, it says: In the United States, African Americans have the highest rate of hypertension when compared to European Americans, Asian Americans, and Latin Americans, due to a combination of environmental and genetic factors. This sentence provides a bit more context, though not as much as described in this current document.
Brown MJ. Hypertension and ethnic group. BMJ. 2006 Apr 8;332(7545):833-6. doi: 10.1136/bmj.332.7545.833. Erratum in: BMJ. 2006 May 13;332(7550):1138. PMID: 16601044; PMCID: PMC1432176.
Campos CL, Rodriguez CJ. High blood pressure in Hispanics in the United States: a review. Curr Opin Cardiol. 2019 Jul;34(4):350-358. doi: 10.1097/HCO.0000000000000636. PMID: 31045586.
Clark R, Anderson NB, Clark VR, Williams DR. Racism as a stressor for African Americans. A biopsychosocial model. Am Psychol. 1999 Oct;54(10):805-16. doi: 10.1037//0003-066x.54.10.805. PMID: 10540593.
Cooper RS, Wolf-Maier K, Luke A, Adeyemo A, Banegas JR, Forrester T, Giampaoli S, Joffres M, Kastarinen M, Primatesta P, Stegmayr B, Thamm M. An international comparative study of blood pressure in populations of European vs. African descent. BMC Med. 2005 Jan 5;3:2. doi: 10.1186/1741-7015-3-2. PMID: 15629061; PMCID: PMC545060.
Elfassy T, Zeki Al Hazzouri A, Cai J, Baldoni PL, Llabre MM, Rundek T, Raij L, Lash JP, Talavera GA, Wassertheil-Smoller S, Daviglus M, Booth JN 3rd, Castaneda SF, Garcia M, Schneiderman N. Incidence of Hypertension Among US Hispanics/Latinos: The Hispanic Community Health Study/Study of Latinos, 2008 to 2017. J Am Heart Assoc. 2020 Jun 16;9(12):e015031. doi: 10.1161/JAHA.119.015031. Epub 2020 Jun 1. PMID: 32476602; PMCID: PMC7429033.
Fuchs FD. Why do black Americans have higher prevalence of hypertension?: an enigma still unsolved. Hypertension. 2011 Mar;57(3):379-80. doi: 10.1161/HYPERTENSIONAHA.110.163196. Epub 2011 Feb 7. PMID: 21300666
Graves JL. 2005a. The Emperor’s New Clothes: Biological Theories of Race at the Millennium (New Brunswick NJ: Rutgers University Press)
Graves, JL. 2005b. The Race Myth: Why We Pretend Race Exists in America. Dutton Books: New York City
Hicken MT, Lee H, Morenoff J, House JS, Williams DR. Racial/ethnic disparities in hypertension prevalence: reconsidering the role of chronic stress. Am J Public Health. 2014 Jan;104(1):117-23. doi: 10.2105/AJPH.2013.301395. Epub 2013 Nov 14. PMID: 24228644; PMCID: PMC3910029.
Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: analysis of worldwide data. Lancet. 2005 Jan 15-21;365(9455):217-23. doi: 10.1016/S0140-6736(05)17741-1. PMID: 15652604
Krieger N, Sidney S. Racial discrimination and blood pressure: the CARDIA Study of young black and white adults. Am J Public Health. 1996 Oct;86(10):1370-8. doi: 10.2105/ajph.86.10.1370. PMID: 8876504; PMCID: PMC1380646.
Lujan HL, DiCarlo SE. The "African gene" theory: it is time to stop teaching and promoting the slavery hypertension hypothesis. Adv Physiol Educ. 2018 Sep 1;42(3):412-416. doi: 10.1152/advan.00070.2018. PMID: 29972056.
Perneger TV, Klag MJ, Whelton PK. Race and socioeconomic status in hypertension and renal disease. Curr Opin Nephrol Hypertens. 1995 May;4(3):235-9. doi: 10.1097/00041552-199505000-00006. PMID: 7648218.
Read JG, Emerson MO, Tarlov A. Implications of black immigrant health for U.S. racial disparities in health. J Immigr Health. 2005; 7(3): 205-212. doi:10.1007/s10903-005-3677-6
Roberts CB, Vines AI, Kaufman JS, James SA. Cross-sectional association between perceived discrimination and hypertension in African-American men and women: the Pitt County Study. Am J Epidemiol. 2008 Mar 1;167(5):624-32. doi: 10.1093/aje/kwm334. Epub 2007 Dec 13. PMID: 18083714.
Zilbermint M, Hannah-Shmouni F, Stratakis CA. Genetics of Hypertension in African Americans and Others of African Descent. Int J Mol Sci. 2019 Mar 2;20(5):1081. doi: 10.3390/ijms20051081. PMID: 30832344; PMCID: PMC6429313.
Chapter 11: Pulmonary Pathology
Sarcoidosis
A high incidence [of sarcoidosis] in Danish and Swedish populations, and in the United States among African Americans (in whom the frequency is 10 times higher than in whites). (BP10 p 512).
First, I highly recommend the article by Hena (2020). This paper addresses the complexity of addressing social, economic and genetic variables in disease.
Sarcoidosis is a multisystem disease that is characterized by granulomatous inflammation. Onset is typically between the ages of 20 and 40 years. The etiology is unknown, though it appears to be due to an inappropriate immune response in patients with a genetic susceptibility. The most common presentations include bilateral hilar lymphadenopathy and involvement of the lung parenchyma (about 90% of cases). Eye and skin lesions are the next most common manifestation.
A genetic component is likely based on familial clustering and correlation with multiple susceptibility loci, such as human leukocyte antigen genes and other genes that relate to antigen processing and presentation (Rybicki BA et al, 2001; Hena KM, 2020). Multiple genes are likely involved in disease susceptibility.
Sarcoidosis is probably a constellation of diseases, not a single disease entity. Different manifestations have been noted in the United States between patients of European descent and those of African descent: as noted by Robbins, the frequency in the latter population is 10 times higher than the former (Baughman RP et al, 2016). Moreover, while both populations are equally prone to develop pulmonary symptoms, patients of African descent more commonly have extrapulmonary manifestations involving skin, bone marrow, eyes, liver and extrapulmonary lymph nodes. In addition, pulmonary disease is often worse in African Americans, likely due to social determinants of health (see below).
Increased disease severity at presentation (i.e., lower FEV1, greater number of organs involved) is associated with lower income, lack of access to private health insurance or Medicare, barriers to care (e.g., delay in getting medications or a doctor’s appointment, concern about cost) (Rabin DL et al, 2004). In this study, low income (<$20,000/year) patients were more likely to be African American, female, have a high school education or less, lack insurance/had public insurance or Medicaid, report barriers to care and miss one or more appointments in a 6-month period. Barriers to care, combined with other issues such as discounting of symptoms in African American patients (as discussed in the pharmacology lecture on disparities in pain management) and delay in seeking medical care due to mistrust in the health profession, may contribute to the worse prognosis of African American patients. African Americans with sarcoidosis are at increased risk for decreased FVC and new organ involvement over time when compared to patients of European descent and have a higher mortality rate (Swigriss JJ et al, 2011; Mirsaedi M et al, 2015).
In the 11th edition, the wording was changed to this: A high incidence in Danish and Swedish populations and in the United States among people of African descent (in whom the frequency is 2 to 3 times higher than in those of European descent).
This alteration is not entirely satisfactory for a number of reasons. For one thing, it suggests that ancestry is the primary driver. There is no evidence that a single gene is responsible for this disease; polygenic diseases have much more complicated inheritance patterns, particularly when one considers population admixture.
Therefore, it is more accurate to refer to an increased incidence in African Americans, since this brings in the matter of social determinants of health. However, even this is problematic: while there is statistical significance (2- to 3-fold difference), the clinical significance of this difference is underwhelming. Sarcoidosis incidence is reported in 17.8 per 100,000 in “Black Americans” and 8.1 per 100,000 in “White Americans” (Baughman RP et al, 2016).
It would have been preferable to state, as we have elsewhere in Robbins, that incidence and severity are related to a combination of genetic and environmental factors and vary according to population.
Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America. Analysis Based on Health Care Use. Ann Am Thorac Soc. 2016 Aug;13(8):1244-52. doi: 10.1513/AnnalsATS.201511-760OC. PMID: 27509154.
Baughman RP, Teirstein AS et al; Case Control Etiologic Study of Sarcoidosis (ACCESS) research group. Clinical characteristics of patients in a case control study of sarcoidosis. Am J Respir Crit Care Med. 2001 Nov 15;164(10 Pt 1):1885-9. doi: 10.1164/ajrccm.164.10.2104046. PMID: 11734441
Hena KM. Sarcoidosis Epidemiology: Race Matters. Front Immunol. 2020 Sep 15;11:537382. doi: 10.3389/fimmu.2020.537382. PMID: 33042137; PMCID: PMC7522309.
Mirsaeidi M, Machado RF, Schraufnagel D, Sweiss NJ, Baughman RP. Racial difference in sarcoidosis mortality in the United States. Chest. 2015 Feb;147(2):438-449. doi: 10.1378/chest.14-1120. PMID: 25188873; PMCID: PMC4314818.
Rabin DL, Thompson B, Brown KM, Judson MA, Huang X, Lackland DT, Knatterud GL, Yeager H Jr, Rose C, Steimel J. Sarcoidosis: social predictors of severity at presentation. Eur Respir J. 2004 Oct;24(4):601-8. doi: 10.1183/09031936.04.00070503. PMID: 15459139.
Rybicki BA, et al. Familial aggregation of sarcoidosis. A case-control etiologic study of sarcoidosis (ACCESS). Am J Respir Crit Care Med. 2001 Dec 1;164(11):2085-91. doi: 10.1164/ajrccm.164.11.2106001. PMID: 11739139.
Swigris JJ, Olson AL, Huie TJ, Fernandez-Perez ER, Solomon J, Sprunger D, Brown KK. Sarcoidosis-related mortality in the United States from 1988 to 2007. Am J Respir Crit Care Med. 2011 Jun 1;183(11):1524-30. doi: 10.1164/rccm.201010-1679OC. Epub 2011 Feb 17. PMID: 21330454; PMCID: PMC3137141.
Pulmonary adenocarcinoma
Roughly 10% of adenocarcinomas in non-Asian persons (and roughly 50% of adenocarcinomas in Asian populations!) express constitutively activating mutations in epidermal growth factor receptor (EGFR).
The “classic patient” with an EGFR mutation is an Asian female who has never smoked and who has an adenocarcinoma. However, as multiple researchers have demonstrated, EGFR mutations are readily identified in Asian men (44%) and smokers (39.3%) (Shi Y et al, 2014; Zhang YL, 2016). Other types of lung cancer (e.g., squamous cell carcinoma and small cell carcinoma) demonstrate EGFR mutations extremely rarely, often due to an admixture of an adenocarcinomatous component.
Initially, EGFR mutations were classified simply as mutant or wildtype, with mutant alleles rendering a tumor susceptible to tyrosine kinase inhibitor (TKI) therapy. We now know that there are multiple different EGFR mutations with variable sensitivities to TKI therapy; these are classified as sensitive, less sensitive and resistant. The survival benefit of identifying mutations is clear: in unselected patients, TKI treatment is associated with a response in 8 – 9% of patients with a median time to progression of 2.2 to 3.0 months whereas in patients with identified mutations, 68% responded with a mean progression free survival of 12 months (Lindemann NI et al, 2013). In addition, there are now multiple generations of TKI drugs and mutation-specific TKI therapy may be more effective than current regimens (Kobayashi Y et al, 2016).
Shi and colleagues found that while their entire “Asian” study population had a 51.4% incidence of EGFR mutation, when broken down by country of origin, mutation incidence ranged from 22.2% in India to 64.2% in Vietnam. Once more, the broad category of “race” simply does not capture the range of the population specific variation. I have compiled a table from a variety of articles. Some of the difference likely relates to the methodology used, the mutations assessed and population admixture. Shi and others have also examined EGFR mutations based on “race”. There is overlap in countries where the populations consists of almost entirely one “race” (e.g., Vietnam).


The real issue here is resources for care: in the United States, EGFR testing is widely available. The College of American Pathologists recommends that “EGFR molecular testing should be used to select patients for EGFR-targeted TKI therapy, and patients with lung adenocarcinoma should not be excluded from testing on the basis of clinical characteristics [e.g., “race”, ethnicity]” (Lindemann NI et al, 2013). In resource-rich countries, so-called “race” and ethnicity should not be the factor that determines if mutational testing is performed. However, in lower income countries where testing may not be universally available, cost-benefit analysis may result in some patients not being tested. Resources also come into play when it comes to actually paying for tyrosine kinase inhibitors which can cost up to $10,000/month in the United States (Olszewski AJ et al, 2018).
In the 11th edition of Robbins, we have changed the terminology to reflect that there is great variability based on population, not race: “The frequency of this mutation varies in different populations.”
Benbrahim Z, Antonia T, Mellas N. EGFR mutation frequency in Middle East and African non-small cell lung cancer patients: a systematic review and meta-analysis. BMC Cancer. 2018 Sep 14;18(1):891. doi: 10.1186/s12885-018-4774-y. PMID: 30217176; PMCID: PMC6137870.
Calibasi-Kocal G, et al. EGFR mutation status in a series of Turkish non-small cell lung cancer patients. Biomed Rep. 2020 Aug;13(2):2. doi: 10.3892/br.2020.1308. Epub 2020 Jun 2. PMID: 32509305; PMCID: PMC7271734.
Douillard JY, Ostoros G, Cobo M, Ciuleanu T, McCormack R, Webster A, Milenkova T. First-line gefitinib in Caucasian EGFR mutation-positive NSCLC patients: a phase-IV, open-label, single-arm study. Br J Cancer. 2014 Jan 7;110(1):55-62. doi: 10.1038/bjc.2013.721. Epub 2013 Nov 21. PMID: 24263064; PMCID: PMC3887309.
Fakhruddin N, Mahfouz R, Farhat F, Tfayli A, Abdelkhalik R, Jabbour M, Yehia L, Mahfoud Z, Zaatari G. Epidermal growth factor receptor and KRAS mutations in lung adenocarcinoma: a retrospective study of the Lebanese population. Oncol Rep. 2014 Nov;32(5):2223-9. doi: 10.3892/or.2014.3406. Epub 2014 Aug 14. PMID: 25120214.
Huntington SF, Davidoff AJ, Gross CP. Precision Medicine in Oncology II: Economics of Targeted Agents and Immuno-Oncology Drugs. J Clin Oncol. 2020 Feb 1;38(4):351-358. doi: 10.1200/JCO.19.01573. Epub 2019 Dec 5. PMID: 31804866.
Imyanitov EN, et al. Distribution of EGFR Mutations in 10,607 Russian Patients with Lung Cancer. Mol Diagn Ther. 2016 Aug;20(4):401-6. doi: 10.1007/s40291-016-0213-4. PMID: 27259329.
Jazieh AR, Jaafar H, Jaloudi M, Mustafa RS, Rasul K, Zekri J, Bamefleh H, Gasmelseed A. Patterns of epidermal growth factor receptor mutation in non-small-cell lung cancers in the Gulf region. Mol Clin Oncol. 2015 Nov;3(6):1371-1374. doi: 10.3892/mco.2015.644. Epub 2015 Sep 16. PMID: 26807249; PMCID: PMC4665377.
Kobayashi Y, Mitsudomi T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspectives for individualized treatment strategy. Cancer Sci. 2016 Sep;107(9):1179-86. doi: 10.1111/cas.12996. Epub 2016 Aug 9. PMID: 27323238; PMCID: PMC5021039.
Lindeman NI, Cagle PT, Beasley MB, Chitale DA, Dacic S, Giaccone G, Jenkins RB, Kwiatkowski DJ, Saldivar JS, Squire J, Thunnissen E, Ladanyi M, College of American Pathologists International Association for the Study of Lung Cancer and Association for Molecular Pathology. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Mol Diagn. 2013 Jul;15(4):415-53. doi: 10.1016/j.jmoldx.2013.03.001. Epub 2013 Apr 4. Erratum in: J Mol Diagn. 2013 Sep;15(5):730. PMID: 23562183.
Lindeman NI, Cagle PT, Aisner DL, Arcila ME, Beasley MB, Bernicker EH, Colasacco C, Dacic S, Hirsch FR, Kerr K, Kwiatkowski DJ, Ladanyi M, Nowak JA, Sholl L, Temple-Smolkin R, Solomon B, Souter LH, Thunnissen E, Tsao MS, Ventura CB, Wynes MW, Yatabe Y. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. Arch Pathol Lab Med. 2018 Mar;142(3):321-346. doi: 10.5858/arpa.2017-0388-CP. Epub 2018 Jan 22. PMID: 29355391.
Mohammadzadeh S, Jowkar Z, Mirzai M, Geramizadeh B. Epidermal Growth Factor Receptor (EGFR) Gene Mutation Analysis in Adenocarcinoma of Lung, the First Report from Iran. Iran J Pathol. 2019 Winter;14(1):1-7. doi: 10.30699/IJP.14.1.1. Epub 2018 Dec 27. PMID: 31531095; PMCID: PMC6708563.
Naderi S, Ghorra C, Haddad F, Kourie HR, Rassy M, El Karak F, Ghosn M, Abadjian G, Kattan J. EGFR mutation status in Middle Eastern patients with non-squamous non-small cell lung carcinoma: A single institution experience. Cancer Epidemiol. 2015 Dec;39(6):1099-102. doi: 10.1016/j.canep.2015.08.016. Epub 2015 Sep 9. PMID: 26362141.
Olszewski AJ, Zullo AR, Nering CR, Huynh JP. Use of Charity Financial Assistance for Novel Oral Anticancer Agents. J Oncol Pract. 2018 Apr;14(4):e221-e228. doi: 10.1200/JOP.2017.027896. Epub 2018 Feb 13. PMID: 29443649; PMCID: PMC5951296.
Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, Majem M, Lopez-Vivanco G, Isla D, Provencio M, Insa A, Massuti B, Gonzalez-Larriba JL, Paz-Ares L, Bover I, Garcia-Campelo R, Moreno MA, Catot S, Rolfo C, Reguart N, Palmero R, Sánchez JM, Bastus R, Mayo C, Bertran-Alamillo J, Molina MA, Sanchez JJ, Taron M; Spanish Lung Cancer Group. Screening for epidermal growth factor receptor mutations in lung cancer. N Engl J Med. 2009 Sep 3;361(10):958-67. doi: 10.1056/NEJMoa0904554. Epub 2009 Aug 19. PMID: 19692684.
Shi Y, et al. A prospective, molecular epidemiology study of EGFR mutations in Asian patients with advanced non-small-cell lung cancer of adenocarcinoma histology (PIONEER). J Thorac Oncol. 2014 Feb;9(2):154-62. doi: 10.1097/JTO.0000000000000033. PMID: 24419411; PMCID: PMC4132036.
Zhang YL, Yuan JQ, Wang KF, Fu XH, Han XR, Threapleton D, Yang ZY, Mao C, Tang JL. The prevalence of EGFR mutation in patients with non-small cell lung cancer: a systematic review and meta-analysis. Oncotarget. 2016 Nov 29;7(48):78985-78993. doi: 10.18632/oncotarget.12587. PMID: 27738317; PMCID: PMC5346692.
Chapter Ten: Hematopathology
Various anemias: sickle cell, thalassemia, G6PD deficiency
All of these are likely related, in some way, as evolutionary adaptations that confer a protective effect against malaria. According to Kariuki and colleagues. over the last 5,000 years, malaria has been the greatest contributor to childhood mortality. In the World Malaria report by the WHO, it was noted that in 2019, it was estimated that more than 400,00 people died from malaria. This suggests that there still is significant selection pressure on variants that protect against malaria.
Kariuki SN, Williams TN. Human genetics and malaria resistance. Hum Genet. 2020 Jun; 139(6-7): 801-811. doi: 10.1007/s00439-020-02142-6. Epub 2020 Mar 4. PMID: 32130487; PMCID: PMC7271956.
In the United States, approximately 8% of blacks are heterozygous HbS carriers and about 1 in 600 have sickle cell anemia. (BP10, p 445; EP1, p 139)
There are multiple hemoglobinopathies that we will discuss in week 14 that are related to a heterozygote advantage resulting from resistance to falciparum malaria. While it may be useful to recognize that patients of Western/Central African descent are more likely to have sickle cell disease/trait compared to those of northern European descent, it is also important to recognize that, depending on the area of African ancestry, individuals may have a minimal risk of sickle cell disease/trait. In areas where malaria is not endemic, there was no selective pressure for sickle cell disease/trait. The high incidence in African Americans stems in part from the fact that enslaved individuals were brought to the Americas from areas (left hand figure; doi:10.1016/j.gene.2010.07.008) that had a high incidence of malaria and sickle cell disease/trait (right hand figure). However, there are also regions in Europe, such as Greece, where the frequency of the sickle cell allele is higher than that of African Americans or Southern Africans (Penmen et al. 2012). Similarly, the Yemeni population displays higher frequencies of this trait than those observed in African Americans (Al-Ghazaly et al. 2012.)

Sickle cell disease is the prototypical entity for which a mutation is linked to an evolutionary advantage: heterozygote carriers of the b globin E6V mutation have improved survival/reproductive success in the setting of malaria infection. However, while heterozygotes are protected against malaria, homozygosity is associated with significant morbidity and mortality from sickle cell disease. Eight to 10% of African Americans are heterozygous for HbS and between 70,000 and 100,000 individuals are homozygotes. This figure is lower than the frequencies for sickle cell disease/trait in the western/central Africans populations that African Americans descended from, indicating that these traits are not beneficial in the absence of malaria.
Although the mechanism for increased resistance to malaria is not completely understood, two mechanisms have been proposed: 1) cells infected with P. falciparum tend to sickle more easily than noninfected cells, leading to their destruction and removal from the circulation (Luzzatto L et al, 1970) and 2) impaired parasite growth in the setting of decreased oxygenation (Friedman MJ, 1978). The glutamic acid to valine substitution at position 6 (E6V) in the b globin gene may be present in both alleles (sickle cell anemia; SCA) or one allele (sickle cell trait; SCT). Glutamate is negatively charged, polar and hydrophilic whereas valine is nonpolar and hydrophobic. When HbS is deoxygenated, it has the tendency to polymerize which causes a change in shape from a biconcave disc to a sickled shape.
There have been two theories regarding the origin of the sickle cell mutation: 1) a multicentric model, based on restriction fragment length polymorphisms, that describes 5 independent mutations classified as Arab-Indian, Benin, Cameroon, Central African Republic and Senegal and published primarily in the 1980’s and 2) a unicentric model based on whole genome sequencing that proposes a single mutation event 7,300 years ago (Shriner D and Rotimi, CN, 2018).
In the multicentric model, this distribution is based on separate mutation events. According to the unicentric model, however, African ancestry would be basis for all cases of sickle cell disease. Apparently, more data from the unicentric model group is forthcoming. The unicentric model requires that either there was significant P. falciparum infection in humans before the advent of modern agriculture (> 10,000 ybp) and the allele was at low frequency in the original human populations that dispersed from Eastern Africa (~100,000 YBP) or that there was sufficient recent gene flow from the original mutational source to other regions after 7,300 YBP.
Finally, it is also important to recognize that sickle cell disease/trait is not just a disease seen in individuals of African descent. According to the CDC, “Sickle cell disease (SCD) affects millions of people throughout the world and is particularly common among those whose ancestors came from sub-Saharan Africa; Spanish-speaking regions in the Western Hemisphere (South America, the Caribbean, and Central America); Saudi Arabia; India; and Mediterranean countries such as Turkey, Greece, and Italy.” However, the operational cause of elevated sickle cell frequency is not “geography” per se, but rather ancestry in regions with high transmission of falciparum malaria.
In the 11th edition of Robbins, we emphasized that the sickle cell allele can be found in multiple populations. We also used the appropriate terminology (ancestry) as opposed to socially defined race (African American): The presence of HbS is protective against falciparum malaria and because of this selective pressure the HbS allele is prevalent in areas where malaria was (and, in some instances, still is) endemic, including equatorial Africa and parts of India, southern Europe, and the Middle East. In the United States, approximately 8% of people of African descent are heterozygous HbS carriers, and about 1 in 600 have sickle cell anemia.
Al-Ghazaly J, Al-Dubai W, Abdullah M, Al-Mahagri A, Al-Gharasi L. Characteristics of sickle cell anemia in Yemen. Hemoglobin. 2013;37(1):1-15. doi: 10.3109/03630269.2012.751033. Epub 2012 Dec 12. PMID: 23234436
Baker C, Powell J, Le D, Creary MS, Daley LA, McDonald MA, Royal CD. Implementation of the NCAA Sickle Cell Trait Screening Policy: A Survey of Athletic Staff and Student-athletes. J Natl Med Assoc. 2018 Dec;110(6):564-573. doi: 10.1016/j.jnma.2018.03.004. Epub 2018 Apr 7. PMID: 30129496.
Bunn HF. The triumph of good over evil: protection by the sickle gene against malaria. Blood. 2013 Jan 3;121(1):20-5. doi: 10.1182/blood-2012-08-449397. Epub 2012 Nov 1. PMID: 23118217.
Eaton WA, Bunn HF. Treating sickle cell disease by targeting HbS polymerization. Blood. 2017 May 18;129(20):2719-2726. doi: 10.1182/blood-2017-02-765891. Epub 2017 Apr 6. PMID: 28385699; PMCID: PMC5437829
Gong L, Parikh S, Rosenthal PJ, Greenhouse B. Biochemical and immunological mechanisms by which sickle cell trait protects against malaria. Malar J. 2013 Sep 11;12:317. doi: 10.1186/1475-2875-12-317. PMID: 24025776; PMCID: PMC3847285
Luzzatto L, Nwachuku-Jarrett ES, Reddy S. Increased sickling of parasitised erythrocytes as mechanism of resistance against malaria in the sickle-cell trait. Lancet. 1970 Feb 14;1(7642):319-21. doi: 10.1016/s0140-6736(70)90700-2. PMID: 4189578.
Pagnier J, Mears JG, Dunda-Belkhodja O, Schaefer-Rego KE, Beldjord C, Nagel RL, Labie D. Evidence for the multicentric origin of the sickle cell hemoglobin gene in Africa. Proc Natl Acad Sci U S A. 1984 Mar;81(6):1771-3. doi: 10.1073/pnas.81.6.1771. PMID: 6584911; PMCID: PMC345002.
Penman BS, Gupta S, Buckee CO. The emergence and maintenance of sickle cell hotspots in the Mediterranean. Infect Genet Evol. 2012 Oct;12(7):1543-50. doi: 10.1016/j.meegid.2012.06.001. Epub 2012 Jun 13. PMID: 22704979; PMCID: PMC3438445.
Serjeant GR, Ghosh K, Patel J. Sickle cell disease in India: A perspective. Indian J Med Res. 2016 Jan;143(1):21-4. doi: 10.4103/0971-5916.178582. PMID: 26997009; PMCID: PMC4822363.
Shriner D, Rotimi CN. Whole-Genome-Sequence-Based Haplotypes Reveal Single Origin of the Sickle Allele during the Holocene Wet Phase. Am J Hum Genet. 2018 Apr 5;102(4):547-556. doi: 10.1016/j.ajhg.2018.02.003. Epub 2018 Mar 8. PMID: 29526279; PMCID: PMC5985360.
Whitten CF. Sickle-cell programming--an imperiled promise. N Engl J Med. 1973 Feb 8;288(6):318-9. doi: 10.1056/NEJM197302082880612. PMID: 4682674.
The Thalassemias
The mutations that cause thalassemia are particularly common in Mediterranean, African, and Asian regions in which malaria is endemic. As with HbS, it is hypothesized that globin mutations associated with thalassemia protect against falciparum malaria.
Thalassemias are hereditary blood disorders whose etiology is due to an imbalance in the number of alpha and beta globin chains in the red cell. Hemoglobin tetramers are typically composed of two alpha chains and two beta chains; reduction in the number of either results in thalassemia. The thalassemias are divided into alpha thalassemia, in which there is a reduction in the number of alpha chains, and beta thalassemia, which is due to a reduction in beta chains. These two large categories are further subdivided based on disease severity. The issue is additionally complicated by the fact that a given patient may be a compound heterozygote with more than one mutant allele. Mutations may be in the globin gene, in other elements that regulate globin synthesis or in additional loci that modulate the severity of the disease (Weather DJ, 2001).
The thalassemias originated in equatorial areas of the world where malaria is or was endemic: sub-Saharan Africa, the Middle East, parts of India, China, Southeast Asia and the Mediterranean region (Harteveld CL et al, 2010; Galanello R et al, 2010). Due to population migration and admixture, the thalassemias now have a worldwide distribution, including northern Europe and North and South America, areas where they previously were not seen.
From Weatherall DJ, 2001:
Figure 5 | The global distribution of the α-thalassaemias. Note that the α-thalassaemia distribution is not as well charted as the thalassaemia distribution shown in FIG. 3, and it is therefore not possible to be as precise about the ranges of the various alleles.



However, with the help of Dr. Joseph Graves, Jr., we were able to elaborate in greater detail in the section on chronic renal disease: Chronic kidney disease is about five times more common in African Americans than in European Americans. Recently, polymorphisms in the APOL1 gene have been identified that increase the risk of kidney disease and also confer resistance to trypanosomiasis, suggesting that these alleles arose because of selection pressure in sub-Saharan regions where trypanosome infection is endemic. Although how APOL1 contributes to resistance to the parasite infection or tokidney disease is unknown, these findings have led to clinical trials of APOL1 inhibitors in patients with renal disease.
Beckerman P, Susztak K. APOL1: The Balance Imposed by Infection, Selection, and Kidney Disease. Trends Mol Med. 2018 Aug;24(8):682-695. doi: 10.1016/j.molmed.2018.05.008. Epub 2018 Jun 7. PMID: 29886044; PMCID: PMC6101980.
Collins FS, Doudna JA, Lander ES, Rotimi CN. Human Molecular Genetics and Genomics - Important Advances and Exciting Possibilities. N Engl J Med. 2021 Jan 7;384(1):1-4. doi: 10.1056/NEJMp2030694. Epub 2021 Jan 2. PMID: 33393745.
Conley AB, Rishishwar L, Norris ET, Valderrama-Aguirre A, Mariño-Ramírez L, Medina-Rivas MA, Jordan IK. A Comparative Analysis of Genetic Ancestry and Admixture in the Colombian Populations of Chocó and Medellín. G3 (Bethesda). 2017 Oct 5;7(10):3435-3447. doi: 10.1534/g3.117.1118. PMID: 28855283; PMCID: PMC5633392
Cooper A, Ilboudo H, Alibu VP, Ravel S, Enyaru J, Weir W, Noyes H, Capewell P, Camara M, Milet J, Jamonneau V, Camara O, Matovu E, Bucheton B, MacLeod A. APOL1 renal risk variants have contrasting resistance and susceptibility associations with African trypanosomiasis. Elife. 2017 May 24;6:e25461. doi: 10.7554/eLife.25461. PMID: 28537557; PMCID: PMC5495568.
Dummer PD, Limou S, Rosenberg AZ, et al. APOL1 Kidney Disease Risk Variants: An Evolving Landscape. Semin Nephrol. 2015;35(3):222-236. doi:10.1016/j.semnephrol.2015.04.008
Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, Bernhardy AJ, Hicks PJ, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010 Aug 13;329(5993):841-5. doi: 10.1126/science.1193032. Epub 2010 Jul 15. PMID: 20647424; PMCID: PMC2980843.
Robinson TW, Freedman BI. The Impact of APOL1 on Chronic Kidney Disease and Hypertension. Adv Chronic Kidney Dis. 2019 Mar;26(2):131-136. doi: 10.1053/j.ackd.2019.01.003. PMID: 31023447; PMCID: PMC6601639.
Thomson R, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci U S A. 2014 May 20;111(20):E2130-9. doi: 10.1073/pnas.1400699111. Epub 2014 May 7. PMID: 24808134; PMCID: PMC4034216.
United States Renal Data System. Bethesda, MD: National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases; 2014. Annual Data Report: Epidemiology of Kidney Disease in the United States
Schaefer RT. Racial and Ethnic Groups in the United States 6th Ed. (New York: HarperCollins), 1996.
Syphilis/STIs
A strong racial disparity is evident [in syphilis infection]; African Americans are affected six times more often than whites (BP, p 705)
It is ALWAYS challenging to speak objectively about sexually transmitted infections (STIs). There can be a lot of shame, guilt and embarrassment around the topic of STIs with a tendency to “victim blame” since STIs are related to behaviors (“choices”) such as premarital sex, multiple partners and drug use that do not always match cultural norms. As health providers, we must work to destigmatize STIs and to engage patient trust so that these very treatable diseases are addressed. Societal disparities such as access to care (including issues like fear and distrust of the medical system), access to medication (are there pharmacies in a given neighborhood? are the medications affordable?) and education about STIs also contribute to differences in infection rates between populations.
Syphilis, in particular, has an additional emotional charge due to its association with the horrific Tuskegee experiments on African American men from 1932-1972 (Jones JH,1993).
The statement in Robbins is factually correct: according to the CDC (accessed 2/21/25), syphilis infection rates are about 5 times higher in African Americans and are twice as high in Hispanic men when compared to European American men. You can see the breakdown below by socially defined race and sex.

Community infection rate may be related to residential racial segregation, defined as the spatial distribution of one socially defined race relative to another. Such segregation is particularly common in metropolitan areas and has numerous dimensions: isolation from other racial groups; concentration (increased density) of social, economic and political disadvantage; centralization (most racially segregated areas are in the centers of metropolitan areas associated with poverty and crowding); clustering (contiguous areas of racially segregated areas in metropolitan centers); and unevenness of population distribution in which the population in a community does not reflect that of the urban center as a whole (Biello KB et al, 2013). Of these dimensions, centralization has been shown to correlate most closely with early age at first sexual intercourse. In communities where STI prevalence is higher, due to some of the factors mentioned above, the risk of encountering an infected partner is also higher.
For the 11th edition of Robbins, this race-based association has been removed. The rationale for removing this association is that while the epidemiologic data are correct, they are pejorative and stigmatizing without contributing to good healthcare. A European-American gay male physician pushed back on my statement that, as it stood, the statement was pejorative and stigmatizing, telling me that he and his Bay Area friends and colleagues were very open about their sexual infection history and that these data were not inherently or objectively shameful. Not everyone lives in that world. Furthermore, we should be testing all sexually active individuals for STIs, not making assumptions based on race or ethnicity.
Biello KB, Ickovics J, Niccolai L, Lin H, Kershaw T. Racial differences in age at first sexual intercourse: residential racial segregation and the black-white disparity among U.S. adolescents. Public Health Rep. 2013 Mar-Apr;128 Suppl 1(Suppl 1):23-32. doi: 10.1177/00333549131282S103. PMID: 23450882; PMCID: PMC3562743.
Coker AL, Richter DL, Valois RF, McKeown RE, Garrison CZ, Vincent ML. Correlates and consequences of early initiation of sexual intercourse. J Sch Health. 1994 Nov;64(9):372-7. doi: 10.1111/j.1746-1561.1994.tb06208.x. PMID: 7877279.
Dariotis JK, Sifakis F, Pleck JH, Astone NM, Sonenstein FL. Racial and ethnic disparities in sexual risk behaviors and STDs during young men's transition to adulthood. Perspect Sex Reprod Health. 2011;43(1):51-59. doi:10.1363/4305111
Hamilton DT, Morris M. The racial disparities in STI in the U.S.: Concurrency, STI prevalence, and heterogeneity in partner selection. Epidemics. 2015 Jun;11:56-61. doi: 10.1016/j.epidem.2015.02.003. Epub 2015 Feb 19. PMID: 25979282; PMCID: PMC4435828.United States Centers for Disease Control and Prevention. Sexually transmitted disease surveillance, 2018 https://www.cdc.gov/std/stats18/Syphilis.htm (Accessed on October 17, 2019).
Jones JH. Bad Blood: The Tuskegee Syphilis Experiment 3rd Ed. (New York: The Free Press), 1993.
Sandfort TG, Orr M, Hirsch JS, Santelli J. Long-term health correlates of timing of sexual debut: results from a national US study. Am J Public Health. 2008 Jan;98(1):155-61. doi: 10.2105/AJPH.2006.097444. Epub 2007 Nov 29. PMID: 18048793; PMCID: PMC2156059.